博文
[转载]DESeq2原理不简单
||
上网搜了一下DESeq2软件的工作原理,大体罗列一下,流程可能不是太准确,仅供参考!
不研究不知道,一研究吓一跳,原来DESeq2这么复杂,这10000多的引用量真不是吹的……

DESeq2的差异表达分析涉及多个步骤,具体步骤参见下面流程图中的蓝色部分

简单地说,DESeq2对原始reads进行建模,使用标准化因子(scale factor)来解释库深度的差异。然后,DESeq2估计基因的离散度,并缩小这些估计值以生成更准确的离散度估计,从而对reads count进行建模。最后,DESeq2拟合负二项分布的模型,并使用Wald检验或似然比检验进行假设检验。
1. 建立reads count矩阵
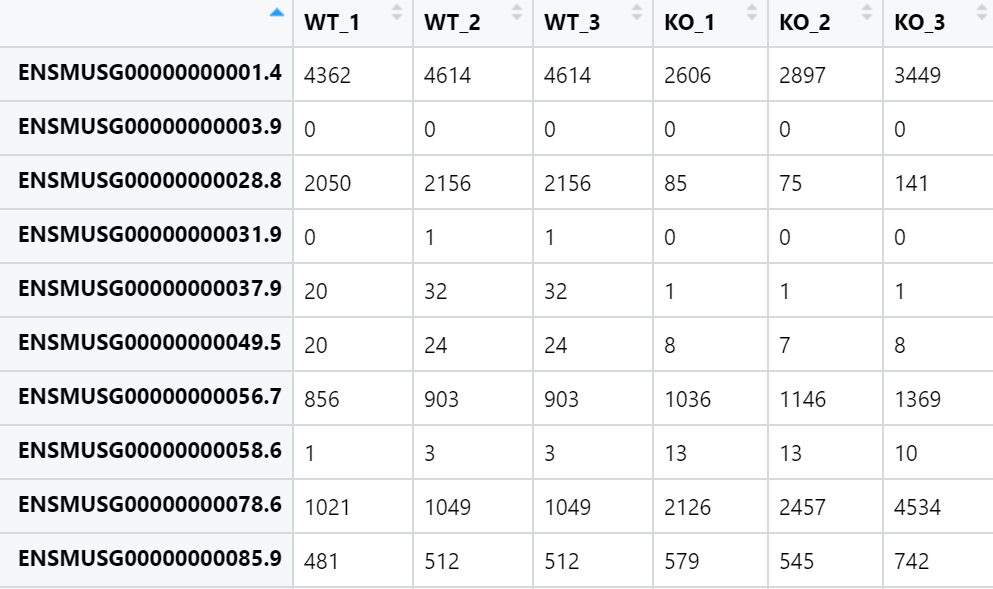
2. 过滤低丰度数据
在独立筛选(independent filtering)中,DESeq2可以去掉在所有样品中平均表达量CPM不大于min.CPM的基因,以减少假阴性。
EdgeR是保留在2个或更多样品中表达量大于min.CPM的基因。
可以尝试不同的cutoff,以获得最佳效果。
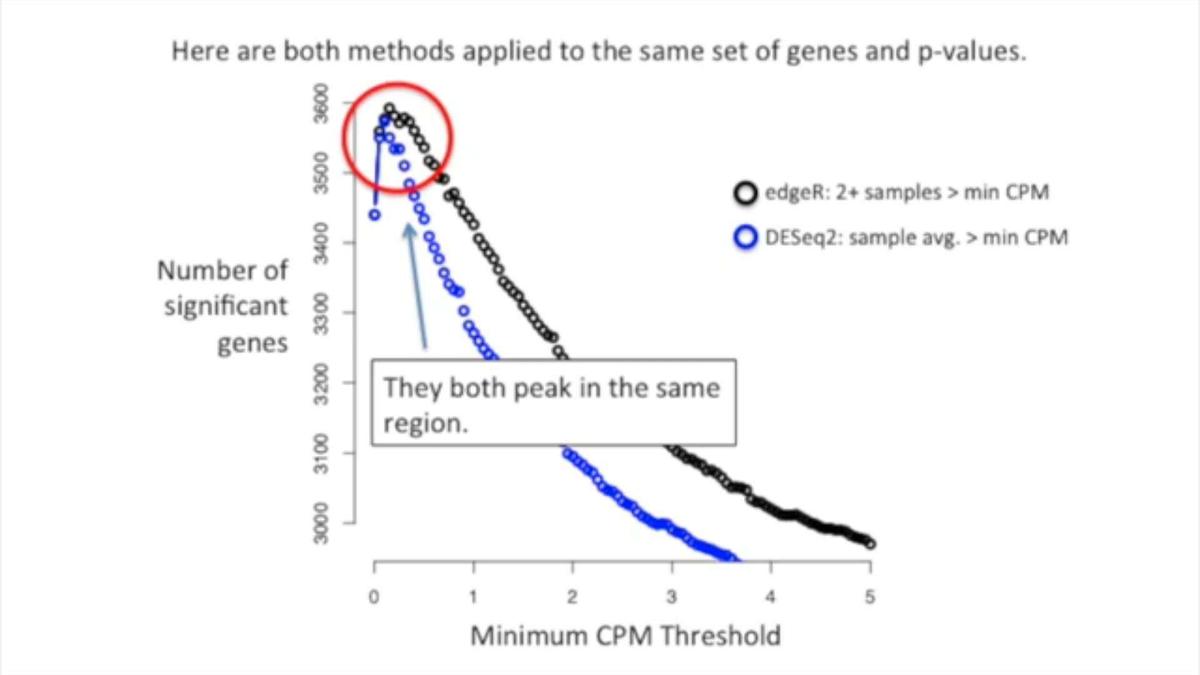
3. 方差稳定变换(variance stabilizing transformation, vst)
vst函数快速估计离散趋势并应用方差稳定变换。该函数从拟合的离散-均值关系中计算方差稳定变换,然后变换count data(除以标准化因子),得到一个近似为同方差的值矩阵(沿均值范围具有恒定的方差)。许多常见的多维数据探索性分析方法,例如聚类或PCA,对于同方差的数据表现良好。数据集小于30个样品可以用rlog,数据集大于30个样品用vst,因为rlog速度慢。
正则化对数变换(regularized-logarithm transformation)
rlog函数将count data转换为log2尺度,以最小化有small counts的行的样本间差异,并使library size标准化。rlog在size factors变化很大的情况下更稳健。
对于RNA-seq raw counts,方差随均值增长。如果直接用size-factor-normalized read counts:counts(dds, normalized=T) 进行主成分分析,结果通常只取决于少数几个表达最高的基因,因为它们显示了样本之间最大的绝对差异。为了避免这种情况,一个策略是采用the logarithm of the normalized count values plus a small pseudocount:log2(counts(dds2, normalized=T) +1)。但是这样,有很低counts的基因将倾向于主导结果。作为一种解决方案,DESeq2为counts数据提供了stabilize the variance across the mean的转换。其中之一是regularized-logarithm transformation or rlog2。对于counts较高的基因,rlog转换可以得到与普通log2转换相似的结果。然而,对于counts较低的基因,所有样本的值都缩小到基因的平均值。
用于绘制PCA图或聚类的数据可以有多种:counts、CPM、log2(counts+1)、log2(CPM+1)、vst、rlog等。
4. 计算归一化系数sizeFactor
通过取log,找中位数,减少异常值对scale factor的影响,从而找到一个合适的scale factor。
将原始的表达量除以每个样本的归一化系数,就得到了归一化之后的表达量。read counts/sizeFactor。
5. 估计基因的离散程度(Estimate gene-wise dispersion)
DESeq2假定基因的表达量符合负二项分布,有两个关键参数,总体均值和离散程度α值。这个α值衡量的是均值和方差之间的关系。
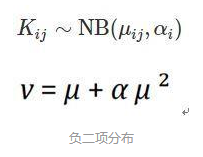
在RNA-seq的reads count数据中,我们需要知道两点:
为了确定差异表达的基因,我们需要根据组内(重复之间)的方差来确定基因的表达值在组间是否有显著差异
组内(重复之间)的变异需要考虑到方差随表达量的平均值增加的情况,如下图所示(每个黑点是一个基因)。
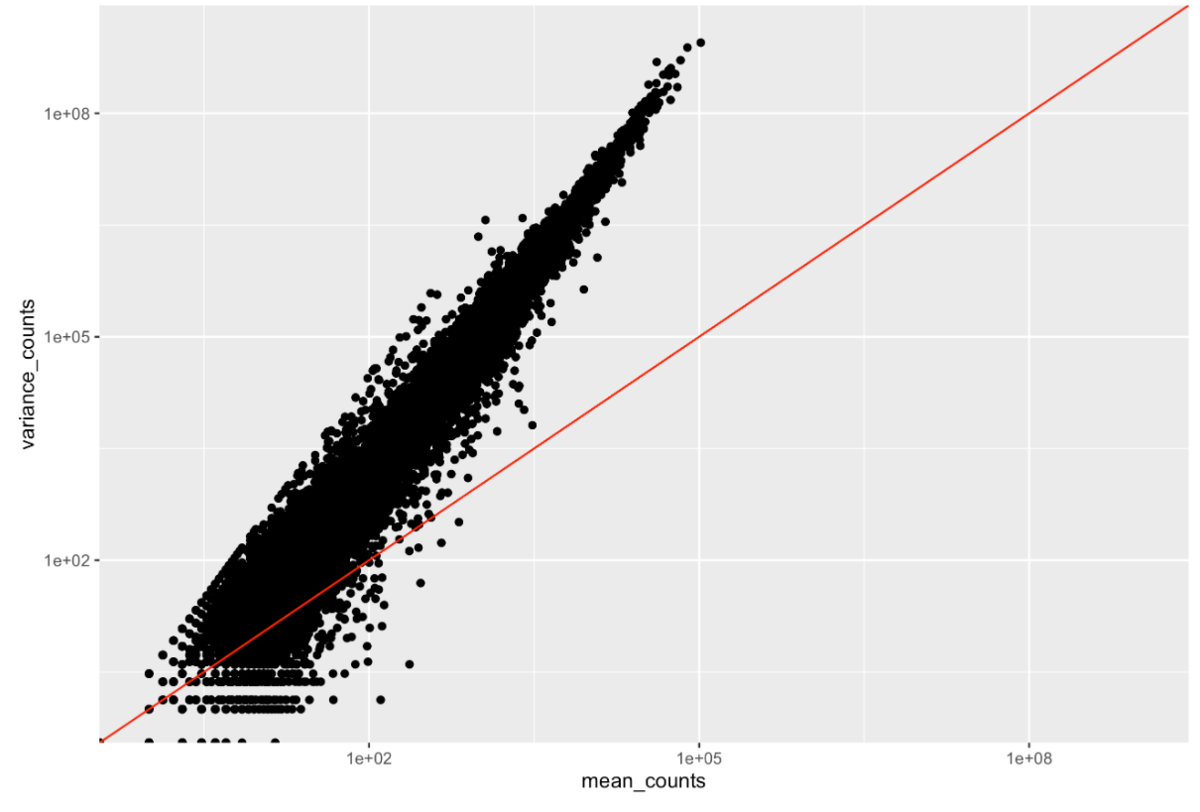
为了更加准确的确定差异表达基因,DESeq2需要解释方差和均值的关系。从上图可知,在低表达的基因中,它们的方差也更低,因此DESeq2不希望差异表达基因都是低表达基因。
DESeq2使用离散度(dispersion)作为方差的度量方式,离散度既可以解释基因表达值的方差也可以解释基因的平均表达值。其具体公式为:Var = μ + α*μ^2。其中Var表示方差,μ表示均值,α表示离散度。因此我们可以得到这么一个关系
那么在表达水平较高的基因中,离散度的平方根就等于方差系数。其中σ是标准差,μ是平均值。那么α=0.01就意味着,在样本生物重复之间存在着10%的标准差。
表达水平较高时,μ对于公式的影响显著小于μ^2
因此,具有相同平均值的基因的离散度仅根据其方差而存在差异,离散度反映了一个给定平均值的基因表达的差异程度。
那么接下来一个比较重要的问题,就是如何将离散度与我们的模型建立联系呢?为了更精确的为我们的数据建模,我们需要知道每个基因组内方差的精确评估值。
但是,生物这个行当,一般3个重复就了不起了,6个重复就顶破天了。看起来似乎挺多的,但是远远不够,这就导致我们得到的组内方差是相当的不可靠……

针对这个问题,DESeq2使用一种叫作shrinkage(暂且翻译成收缩)的方法,共享基因之间的表达信息,根据基因的表达水平生成更为准确的方差估计。DESeq2假设具有相似表达水平的基因也具有相似的方差。这样DESeq2就可以基于基因的平均表达水平和离散度来评估每个基因的离散度。
6. Fit curve to gene-wise dispersion estimates
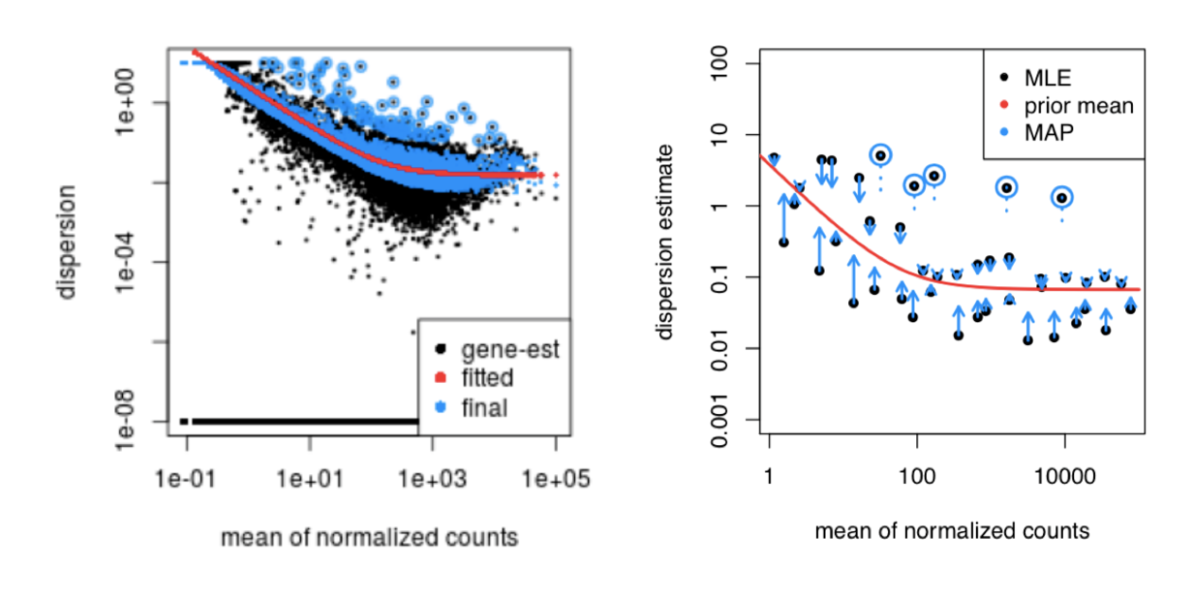
DESeq2的第三步就是根据基因的离散度拟合一个曲线。那么为什么要做拟合呢?不同的基因生物学重复中存在不同的方差,但是,在所有的基因中,将会有一个合理的离散分布。
这个曲线如下图红线所示,其中红线的横坐标是基因的表达强度,纵坐标是理论离散值。而每一个黑点的横坐标是基因的平均表达水平,纵坐标是经过最大似然估计的离散值。
7. Shrink gene-wise dispersion estimates toward the values predicted by the curve
有了拟合曲线,接下来就是对基因表达水平的离散度进行矫正,即:将基因的实际离散度向红线收缩(shrink)。当样本量较小时,该曲线可以让我们更为准确的识别差异表达基因。既然知道要将基因的离散度向红色曲线收缩,那么收缩多少比较合适呢?有两点需要考虑:
a.基因的离散度距离红色曲线的距离
b.样本量(样本量越大,则收缩的越少)
这种方法在差异表达分析时,可以极大的减少数据的假阳性。离散度较低的基因朝着理论值收缩,从而得到一个更为准确的离散值。而那些离散度较高的基因,则不能无脑朝着理论值收缩。
这是因为这类基因可能不遵循建模假设,并且由于生物学或技术原因使得这类基因与其他基因具有更高的方差。DESeq2识别这类基因后,将不采用shrink方法对它们进行处理。下图中蓝色圆圈圈住的便是这类基因。
8. GLM fit for each gene
DESeq2的第五步,对每个基因使用广义线性模型进行拟合,我自己也没搞明白,就不在这里献丑了
9. 差异表达分析
test可以是Wald significance tests或likelihood ratio test(似然比检验),on the difference in deviance between a full and reduced model formula。
获得分析结果,包含6列:baseMean、log2FC、lfcSE、stat、pvalue、padj
baseMean表示所有样本经过归一化系数矫正的read counts(counts/sizeFactor)的均值。log2Foldchange表示该基因的表达发生了多大的变化,是估计的效应大小effect size。对差异表达的倍数取以2为底的对数,变化倍数=2^log2Foldchange。(并不完全相等。log2FC反映的是不同分组间表达量的差异,这个差异由两部分构成,一种是样本间本身的差异,比如生物学重复样本间基因的表达量就有一定程度的差异,另外一部分就是我们真正感兴趣的,由于分组不同或者实验条件不同造成的差异。用归一化之后的数值直接计算出的log2FC包含了以上两种差异,而我们真正感兴趣的只有分组不同造成的差异,DESeq2在差异分析的过程中已经考虑到了样本本身的差异,其最终提供的log2FC只包含了分组间的差异,所以会与手动计算的不同)。
lfcSE(logfoldchange Standard Error)是对于log2Foldchange估计的标准误差估计,效应大小估计有不确定性。
stat是Wald统计量,它是由log2Foldchange除以标准差所得。
pvalue和padj分别代表原始的p值以及经过校正后的p值。adjusted p value less than 0.1 should contain no more than 10% false positives.
Need to filter on adjusted p-values, not p-values, to obtain FDR control. 10% FDR is common because RNA-seq experiments are often exploratory and having 90% true positives in the gene set is ok.
转载自
https://www.jianshu.com/p/bf3fdd21153e
https://www.jianshu.com/p/2689e9a1d10c
https://blog.sciencenet.cn/blog-3431904-1308379.html
上一篇:[转载]简单明了认识箱线图
下一篇:教你怎么查基因启动子和转录因子结合位点