博文
FitHiC V1算法解析(二)
|||
FitHiC V1主要用于识别中程顺式互作
本文接着上一篇博文《FitHiC V1算法解析(一)》继续探讨FitHiC V1的算法过程。该博文中介绍了FitHiC的整体思路,但是这种基于假设检验来检测显著互作的方法,早在2010年就已经提出来了[1],该文章与FitHiC引文的通迅作者都是大神William Stafford Noble。那么FitHiC在之前算法的基础上又有哪些新的改进呢?
在FitHiC中,对上篇博文提到过的binned bionormial method,进行了两点改进:
1. 初始样条拟合(Initial spline fit)
将discrete binning过程换成了equal-occupancy binning,再进行样条拟合,这样可以在某个特定的基因组距离(spline-1)上,更加精确地对连接概率(contact probability)进行评估。
1.1样条拟合
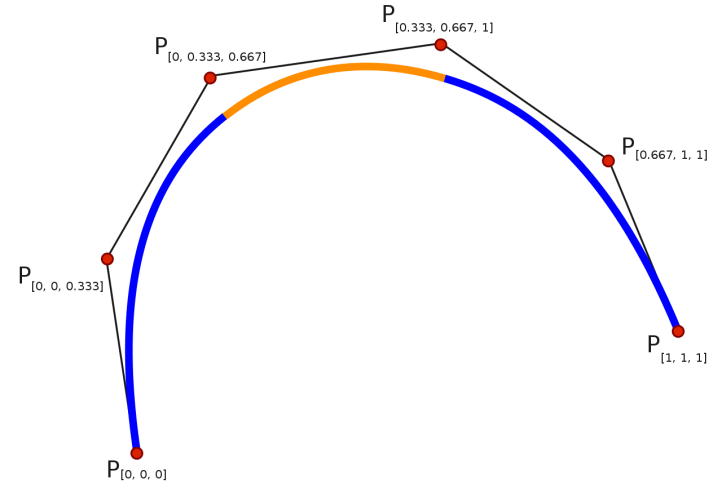
早期工程师制图时,把富有弹性的细长木条(所谓样条)用压铁固定在样点上,在其他地方让它自由弯曲,然后沿木条画下曲线,成为样条曲线。后来用于数学领域,如下图中的的样条函数。图中有6个点P,通过样条函数的拟合,得到一条弧线,即图中蓝色弧线接黄色弧线再接蓝色弧线。
我们还是以上篇博文中讨论过的discrete binning图来说,如下图
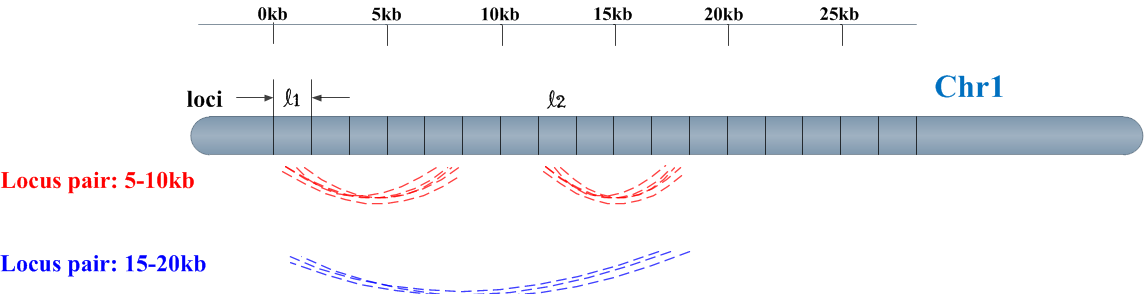
如果两个locus pair中有很多contact距离大约为9kb(或者说平均距离为9kb),而另一组locus pair中有很多contact距离在10.5kb附近,那么按照discrete binning方法,这两组一个分到5kb-10kb,另一个应该会到10-15kb,分开来研究。然而事实上,这两组的差异并不大。这就会产生边限效应(edge effect),而采用样条拟合可以极大的降低这种影响。
另外,FitHiC中改进的方法相比之前Duan[1]提出的discrete binning方法,在处理基因组上较长距离的locus pairs时效果更好。因为较长距离的locus pairs上contact counts比较少,如果采用discrete binning方法则会产生很多空的bin(或contact counts非常少的bin)。
两者比较,图形化展示如下:
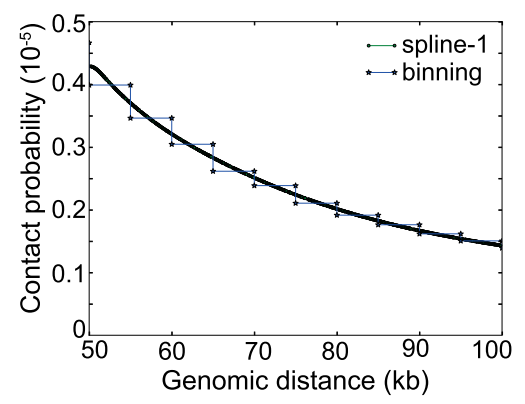
图中binning表示discrete binning方法产生的结果,spline-1表示FitHiC改进的binning方法第一次拟合后的结果
1.2 具体操作
对于顺式互作,求连接概率(contact probability) p时是随机地抽取所有locus pair中的1个,即p=1/M,再将p代入二项分布概率密度公式,如下公式(1)。在FitHiC中则改用了一个以locus pair距离d为自变量的函数,记为f(1)(d)。这个函数具体是怎样的,目前我们还不知道的,只知道它是一个样条函数,我们需要利用观测到的实际数据来拟合出来。

1.2.1 equal-occupancy binning
通过1.1节样条函数的介绍,不难发现,在实际问题中我们需要获得尽量平滑的样条函数。为了达到这一目的,FitHiC做了如下binning操作,这个过程称为"equal-occupancy binning":
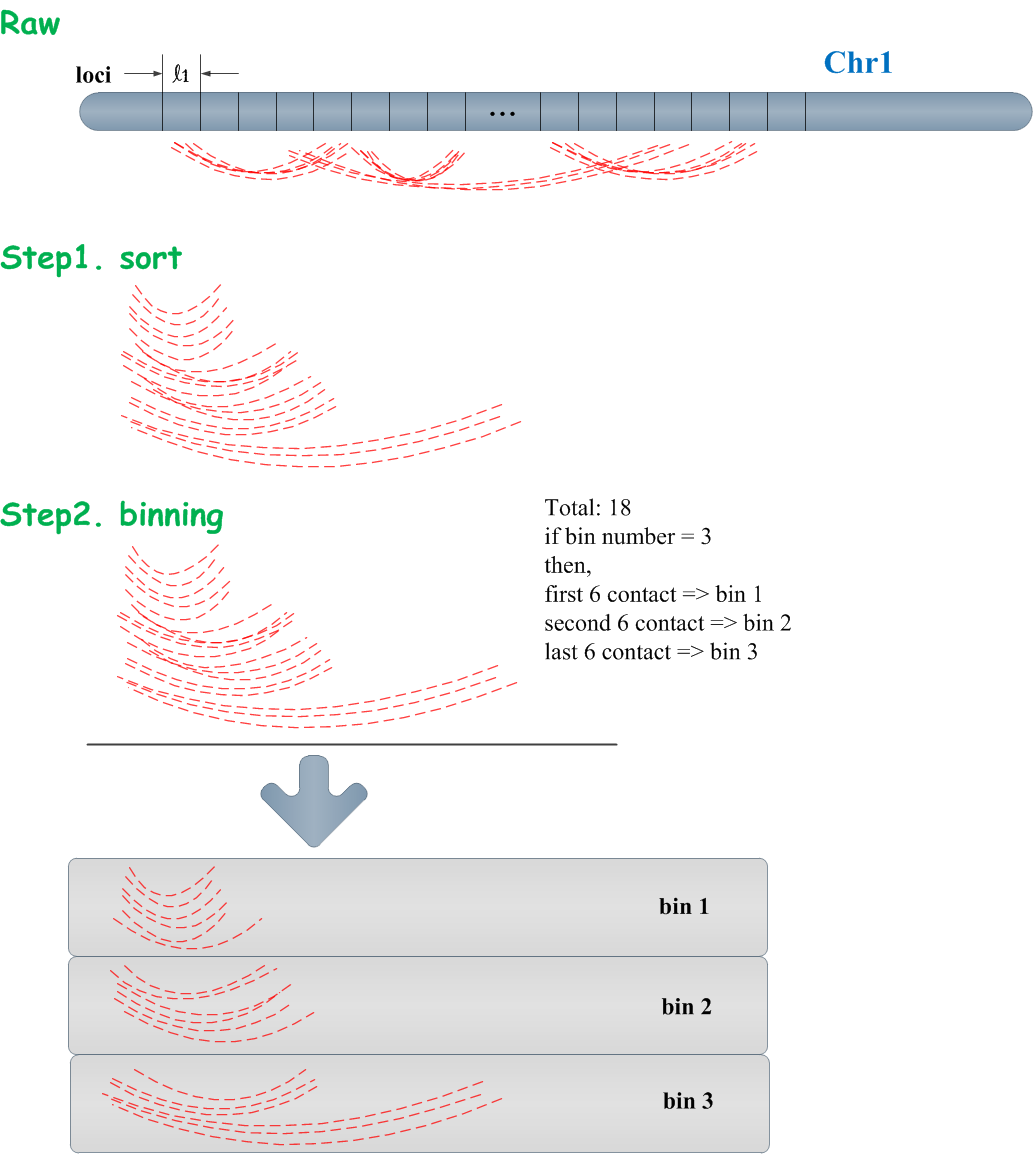
(1) 迭代所有感兴趣的距离范围内(即中等程度范围内)的所有可能的locus pairs,包括那些没有contact的locus pair
(2) 按照基因组上的距离以递增的方式对locus pairs进行排序
(3) 将这些排序后的结果分为b等分(默认b=200)。
注意:这里有一个小bug,即可能相同距离的locus pairs会分到不同的bin中!这个bug在FitHiC V2中得以修复。
这三步具体如下图:
(4) 通过上面equal-occupancy binning的划分方法,划分出b个等分,可以发现第i个等分中可能有多个locus pair,每个locus pair又有很多contact (PE reads)。对等分i计算以下3个值:
A. 第i个等分中,每个locus pair的contact counts平均值。因为分完i个等分后,可以认为每个等分中不同locus pair的contact counts是相同的。
B.从所有mid-range contact中随机挑选一个contact,这个contact来自bin i中某个特定的locus pair的先验概率。这里很关键,但是不太好理解,特别是文章中说的这个先验概率,容易把人绕晕。
打个比方,我们有3个盒子,A盒子装有5个小球,B盒子装有3个小球,C盒子装有2个小球。我们随机抽到了一个小球,请问这个小球来自A盒子的概率。显然为5/(5+3+2)。在这个比方中将小球换成contact,将盒子换为bin i中的locus pair,将三个盒数中所有的小球总数换为所有mid-range contact counts(即N值),再来计算抽到的这个contact来自bin i中某个locus pair的概率,这样就比较好理解了。
C. 第i个等分中所有locus pairs的平均互作距离。
可以通过如下示意图理解这三个值:

1.2.2 spline fitting procedure
这样就获取到了b个点的坐标:
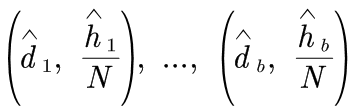
将这些坐标来进行拟合,获取到样条函数f(1)(d)。拟合完后,只要知道locus pair的距离d,代入f(1)(d)就可以算得一个值,这个值即公式(1)中的p
2. 改进零模型(Refining the null model)
FitHiC采用了"二相样条拟合过程(two-phase spline fitting procedure)",在第一相中,我们采用了所有的观测值来拟合初始样条函数f(1)(d)。随后将所有P-value值小于1/M的locus pairs上的contact count屏辟掉(设为0),以去除干扰。其中M值表示locus pairs的总数。这种采用P-value过滤的方法是比较保守的,即使最初的零模型正确,这个过程也平均只会错误地排除约1个非离散值。清理完干扰值后,如前文所述再做一次equal-occupancy binning和spline fitting procedure,这样获取到新的样条函数f(2)(d)可以准确地评估出改进的零模型分布(null distribution)。再计算所有locus pair的P-values和Q-values,包括之前清理掉的离散值。
参考材料:
[1] Zhijun Duan, Mirela Andronescu, Kevin Schutz, Sean McIlwain, Yoo Jung Kim, Choli Lee, Jay Shendure, Stanley Fields, C. Anthony Blau, William S. Noble. A three-dimensional model of the yeast genome. 2010
[2] Ferhat Ay, Timothy L. Bailey, William Stafford Noble. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. 2014
https://blog.sciencenet.cn/blog-2970729-1223477.html
上一篇:FitHiC V1算法解析(一)
下一篇:使用Biopython解析Blast结果(二)