博文
动态网络分析揭示肺癌恶化的关键microRNA
|||
肺癌是全球范围内与癌症相关的人类死亡的主要原因。肺腺癌是最常见的亚型之一,与其它肺癌亚型相比,其基因组变异更为明显。癌症研究中的一个里程碑式发现是非编码RNA的作用,非编码RNA被确定为致癌驱动因子和肿瘤抑制因子。在癌症发展中,非编码RNA在细胞内环境中形成不可分割的RNA水平调节网络的统一体,不同类型RNA之间的动态相互作用和竞争起着至关重要的作用。
我们开发了一种定量的方法,用于在转录后水平上重建肺腺癌演化阶段RNA的相互调控网络。揭示了具有双二分结构的两种特征截然不同的网络,并且发现了在每个阶段都会影响甚至决定患者生存的关键RNA分子。工作建立了更全面的基因数据分析框架,不仅提供了更深入地探索癌症进化机制的复杂网络工具,而且还为研究肺癌生物标志物和潜在药物靶点提供帮助。
近日,相关工作已发表在PLoS Computational Biology, 16(5): e1007793. (2020)。
https://doi.org/10.1371/journal.pcbi.1007793
具体介绍:
Abstract:
Non-coding RNAs are fundamental to the competing endogenous RNA (CeRNA) hypothesis in oncology. Previous work focused on static CeRNA networks. We construct and analyze CeRNA networks for four sequential stages of lung adenocarcinoma (LUAD) based on multi-omics data of long non-coding RNAs (lncRNAs), microRNAs and mRNAs. We find that the networks possess a two-level bipartite structure: common competing endogenous network (CCEN) composed of an invariant set of microRNAs over all the stages and stagedependent, unique competing endogenous networks (UCENs). A systematic enrichment analysis of the pathways of the mRNAs in CCEN reveals that they are strongly associated with cancer development. We also find that the microRNA-linked mRNAs from UCENs have a higher enrichment efficiency. A key finding is six microRNAs from CCEN that impact patient survival at all stages, and four microRNAs that affect the survival from a specific stage. The ten microRNAs can then serve as potential biomarkers and prognostic tools for LUAD.
内容:
非编码RNA(Non-coding RNA, ncRNA)从器官发育到癌症恶化的大量生物过程中都发挥不可或缺的作用。这些RNA 构成了肿瘤学内源性竞争RNA (Competing endogenous RNA, CeRNA)假说的基本组成部分。该假说为探索不同类型癌症的机制和演化提供了复杂网络的研究视角。这个领域之前的工作主要集中在静态CeRNA 网络。癌症的发展过程中,潜在的CeRNA 网络很难保持不变,因此我们研究了CeRNA 网络的动态变化。
将肺腺癌(Lung adenocarcinoma, LUAD)作为一个研究原型案例,对LUAD 四个顺序演化期型,基于长链非编码RNA (Long non-coding RNAs, lncRNAs)、微RNA (microRNA, miRNA)、信使RNA (messenger RNA, mRNA)多组学数据,通过RNA 的差异化表达分析、基于碱基互补配对原理匹配microRNA 靶目标、援引CeRNA 假说选取RNA 之间表达量的负相关方法,来构建四个期型的CeRNA 网络。我们发现网络由microRNAs 及其靶向的mRNAs (或者lncRNAs)组成,具有双二分图结构(Two-level bipartite structure)。
比较LUAD 四期网络中microRNA 的组分,CeRNA 网络自然划分为两类子网络:由全部期型中一组不变的microRNA 构成的共有的内源性竞争网络(The common competing endogenous networks, CCENs),以及不同期型特有的microRNA 构成的独有的内源性竞争网络(The unique competing endogenous networks, UCENs)。CeRNA 网络中mRNA 进行系统的基因本体 (Gene ontology, GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)通路的富集分析,发现构建出的CeRNA 网络与癌 症及其演化相关的通路密切相关,且随着LUAD 期型的恶化与癌症相关程度也更加紧密。另外,UCENs 中microRNA 相连接的mRNAs 相比于从CCENs 或 者UCENs 中直接选取的mRNAs 有更高的富集效率。
具体来说:
1. 基于癌症样本的基因组学数据,采用新颖的数据处理步骤,构建出与LUAD 顺序演化期型相关联的CeRNA网络。从TCGA数据库下载lncRNA、microRNA和mRNA三者的基因表达数据,使用差异化表达分析、miRNA 靶目标匹配、以及CeRNA假设,构建出了癌症四个顺序期型相对应的由三种RNA构成的四期CeRNA网络。
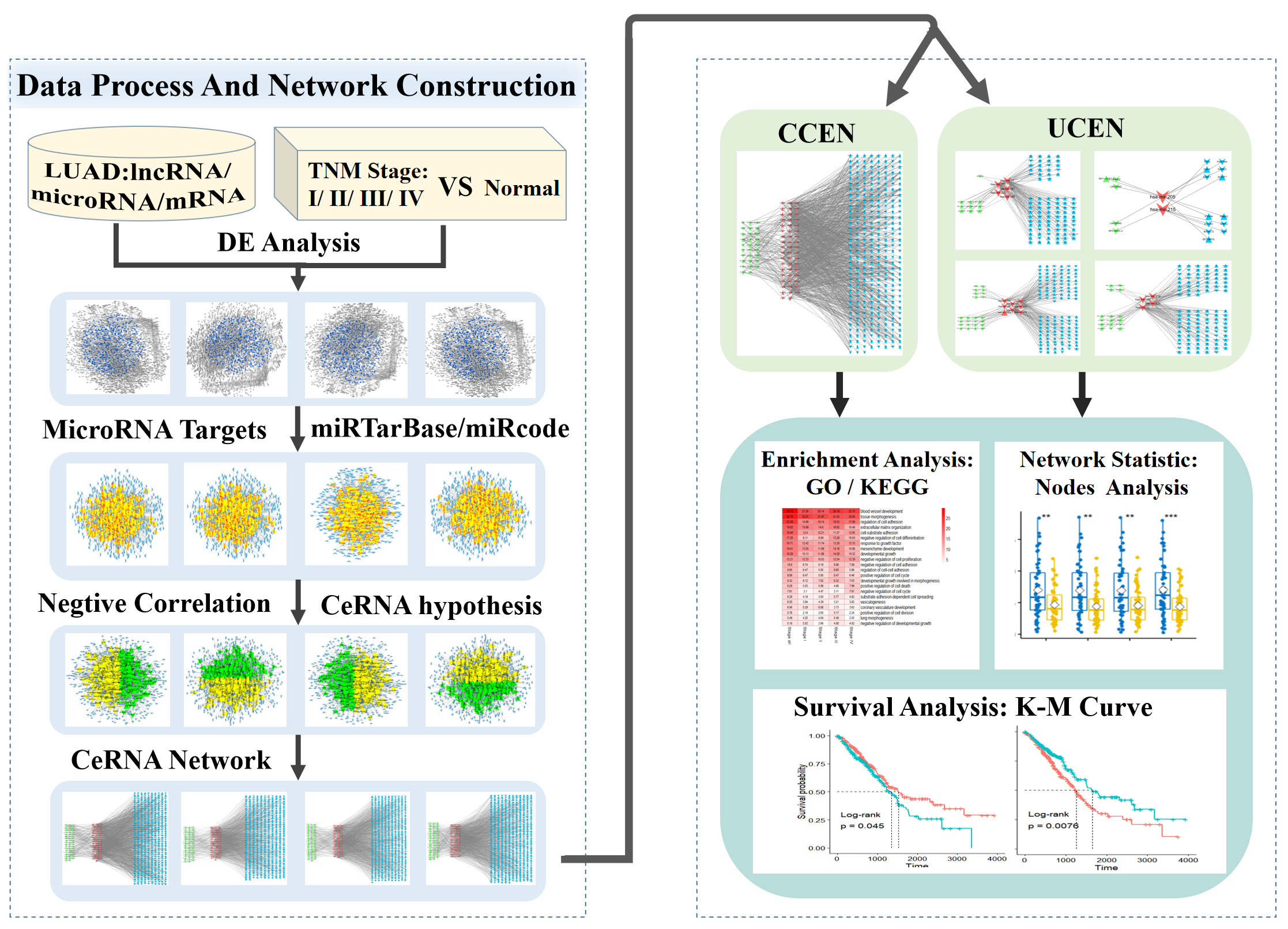
工作流程图
2. 为了验证网络的可靠性,统计了网络中RNA的(FC)值和RNA在样本中的平均表达量。四个期型的LUAD网络中mRNA的GO(Biological process)和KEGG Pathway富集分析,并通过基因富集效率分析,研究了不同网络与癌症相关项目(GO)或通道(KEGG)的相关性。
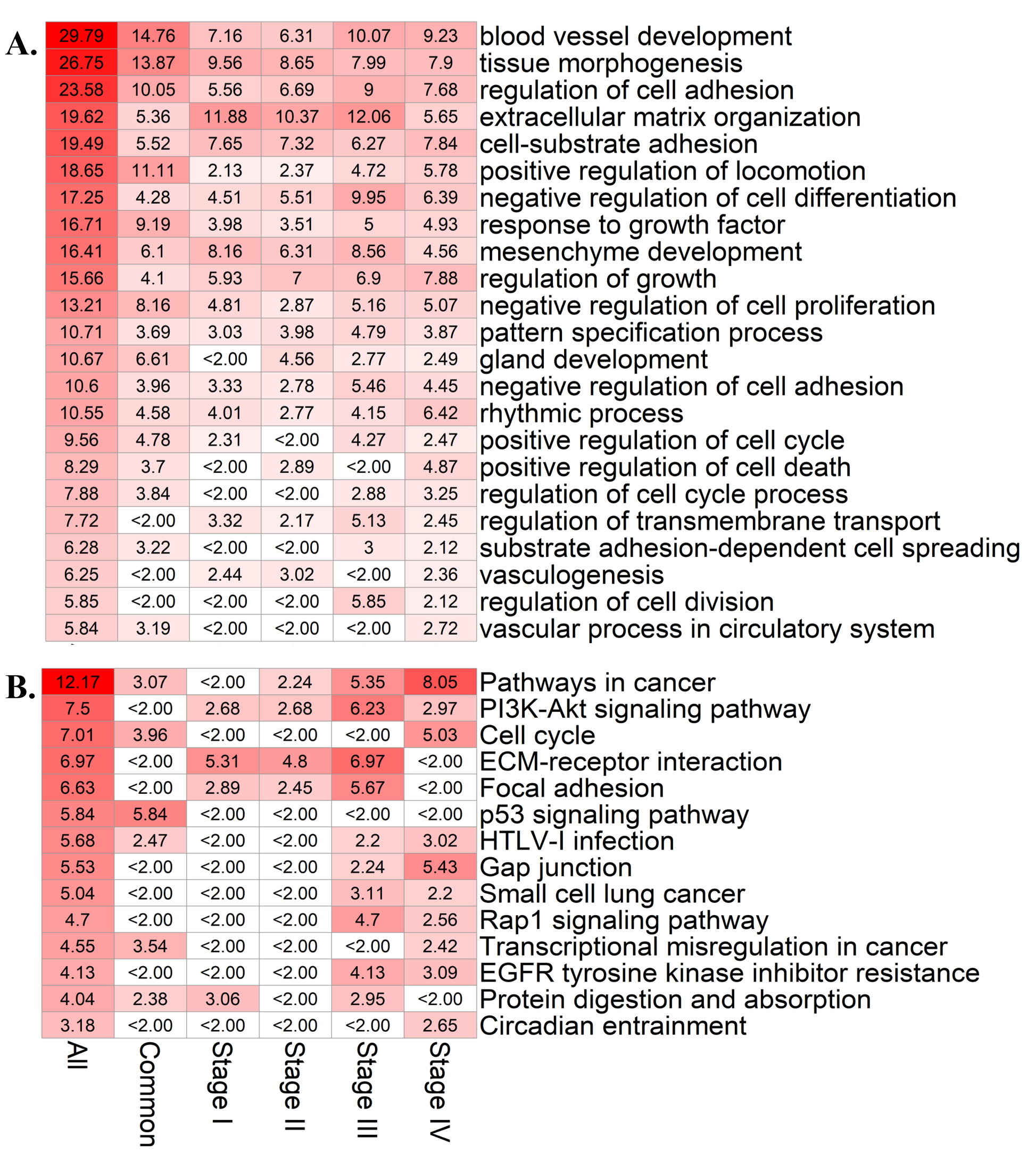
富集分析
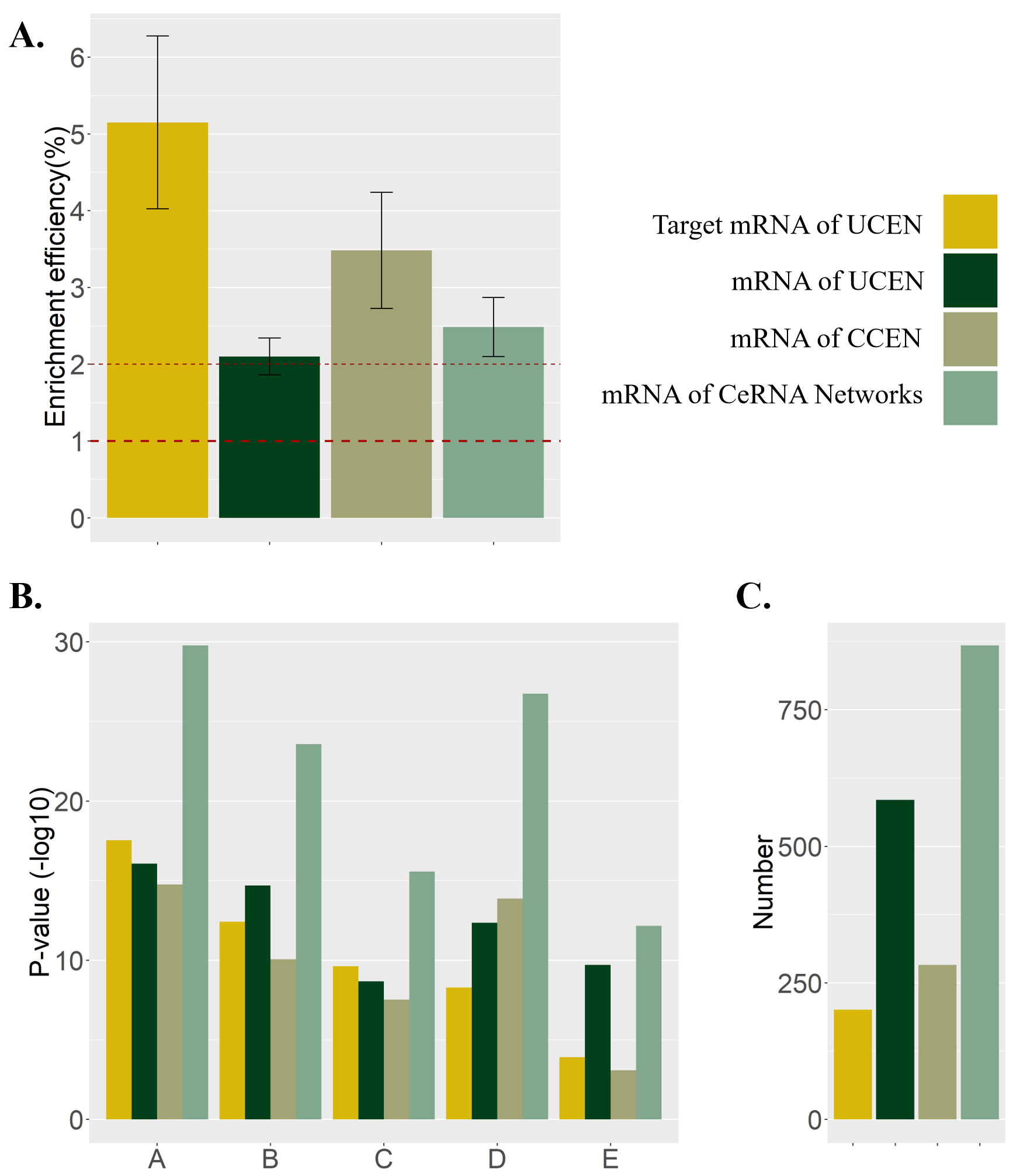
基因富集效率分析
3. 引入CeRAN假说,将构建出的四期CeRNA网络划分为“独有网络”(UCEN)和“共有网络”(CCEN),从网络的视角研究CeRNA分子网络与肺部疾病恶化之间的关系;
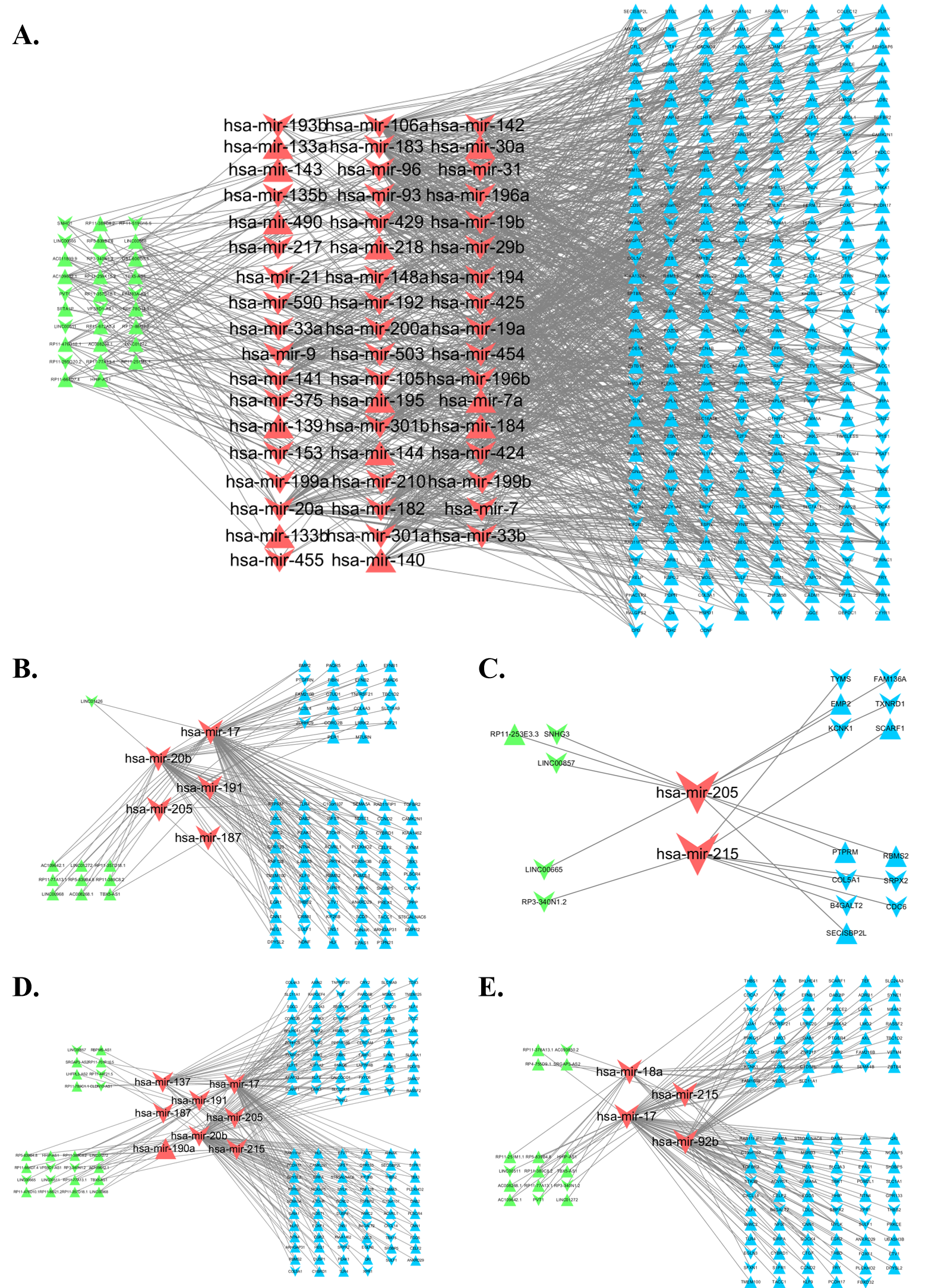
CeRNA网络图
4. 结合病人临床数据,使用K-M生存曲线分析方法研究CeRNA网络中的RNA节点进行对病人生存的影响。
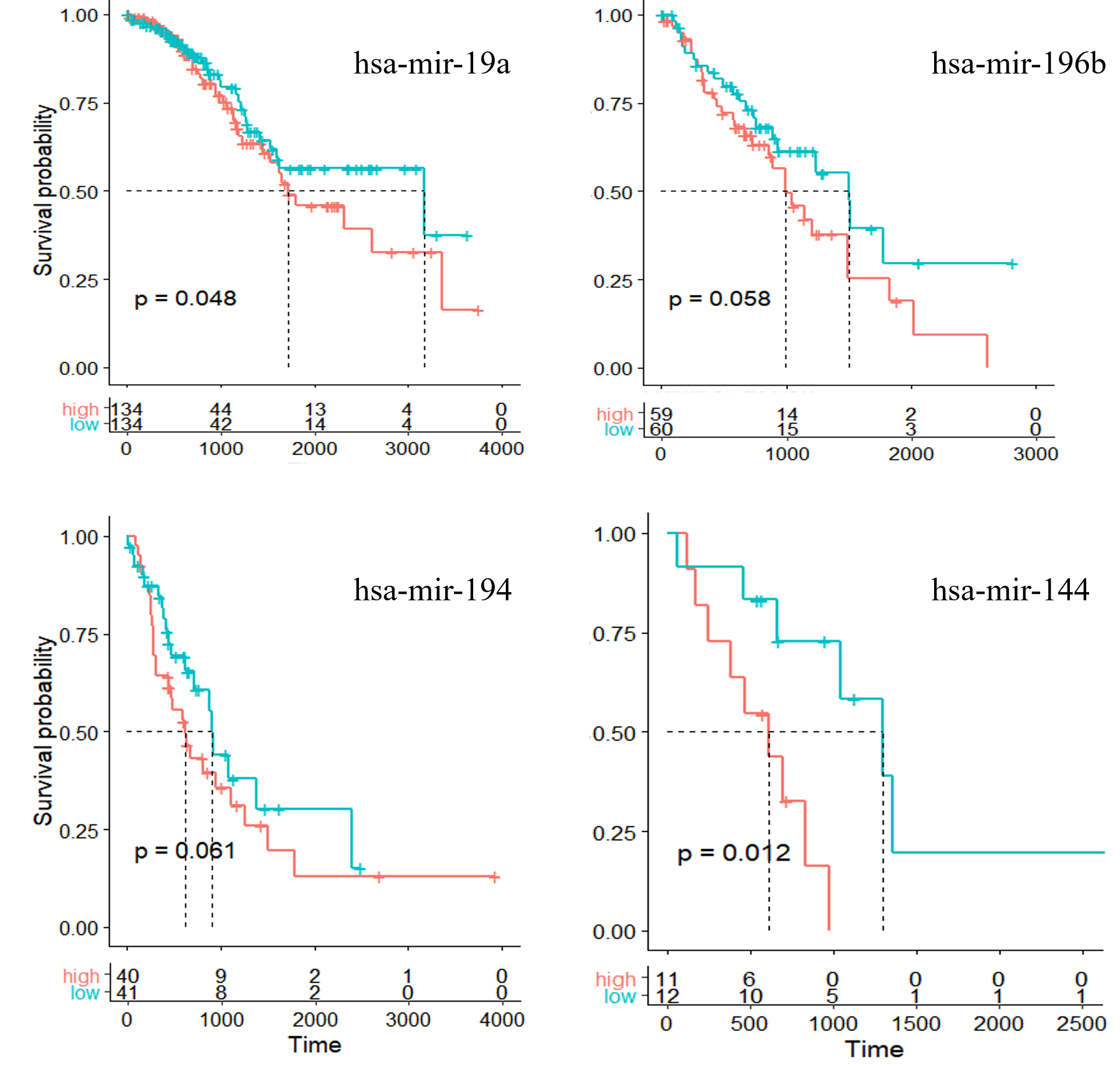
K-M生存曲线分析
创新性:
1. 基于基因组学数据的网络建模方法寻找疾病的生物标志物,从网络角度理解癌症的分子作用机制,并结合生物信息学手段进行验证,研究结果具有一定的可靠性。这为进一步分析网络的性质和探索网络与疾病之间关系奠定基础。
2. 构建出的三种RNA相互作用网络具有双二分图结构,并且网络的节点能够影响癌症恶化过程中样本的生存情况。因而通过构建疾病相关的分子网络能够更好地理解疾病演化规律和寻找潜在的药物靶点。
应用价值:
系统地介绍了疾病相关的基因数据数理流程和网络构建的步骤,并通过对大量的网络节点的统计分析和生物信息相关的第三方数据库进行验证。寻找癌症生物标志物为目前靶向药物的研制和精准医疗指出潜在的方向,为临床应用提供可能的指导。
理论意义:
为复杂网络在生物数据建模方面提供解决思路,其方法框架也可以应用于更加广泛的生物数据分析和基于数据的网络模型构建。这能够极大的扩展复杂网络的应用范围,以及复杂系统理论对其他学科领域的影响,加速交叉学科的快速发展,具有启发意义。
后续扩展工作:
分子生物学、生物信息学相关:
1. 能否将其他种类分子如蛋白质、转录调控因子(TF)等,整合进入现有网络中?;
2. 探寻CeRNA相关的分子作用网络随肺癌期型恶化的演化规律是否在其他疾病中同样存在;
3. 实验验证工作中得到的显著影响LUAD病人的分子标志物在真实生物体内是否存在,并探究其上下游的作用关系;
4. 结合近些年快速发展的单细胞测序技术,研究肺部组织恶化过程中,细胞类型的改变以及不同类型细胞内部分子相互作用网络随癌症恶化过程的演化规律;
复杂网络相关:
5. 应用复杂网络指标和网络算法寻找基因分子网络中普遍存在的规律与疾病之间的关联;
脑科学相关:
6. 随着诸如fMRI、染色荧光标记、微电极阵列等脑相关生物技术的进一步发展,结合新一代测序数据在其他组织器官(如大脑)、甚至其他物种(老鼠、果蝇、恒河猴等)的细胞内进行分子相互作用网络的研究。
硕士论文工作:
本人研究生期间做癌症相关的非编码RNA相互作用网络的研究,涉及复杂网络、生物信息、医学统计等。
孔超. 复杂网络分析揭示肺癌生物标志物[D]. 兰州:兰州大学物理科学与技术学院,2020.
选题的来源和意义:
癌症是主要的公共安全问题之一,并且肺癌在癌症中占比最高。肺癌中,非小细胞癌占有85%,而肺腺癌(LUAD)是非小细胞癌的主要类型。肺腺癌属于原发性癌症,包含更多的基因组变异信息,从而有利于从分子机制角度研究癌症的发生和发展。非编码RNA相互作用,是生物体除中心法则外重要的基因表达调控机制之一,广泛的存在各种组织和器官的细胞时空表达模式中。随着新一代高通量基因组测序技术的发展,研究人员不断发现更多的长链非编码RNA、微RNA、环状RNA等,了解到非编码RNA通过转录后水平的基因调控在生物体内扮演着重要作用。我们的工作中结合了内源性竞争RNA假说,构建RNA分子间相互作用网络,在RNA分子互作用网络层面研究LUAD随癌症恶化的演化规律。
CeRNA网络研究内容:
非编码RNA(Non-coding RNA, ncRNA)从器官发育到癌症恶化的大量生物过程中都发挥不可或缺的作用。这些RNA 构成了肿瘤学内源性竞争RNA(Competing endogenous RNA, CeRNA)假说的基本组成部分。该假说为探索不同类型癌症的机制和演化提供了复杂网络的研究视角。这个领域之前的工作主要集中在静态CeRNA 网络,因此我们研究了CeRNA 网络的动态变化。将肺腺癌(Lung adenocarcinoma, LUAD)作为一个研究原型案例,对LUAD 四个顺序演化期型,基于长链非编码RNA(Long non-coding RNAs, lncRNAs)、微RNA (microRNA, miRNA)、信使RNA (messenger RNA, mRNA)多组学数据,通过RNA 的差异化表达分析、基于碱基互补配对原理匹配microRNA 靶目标、援引CeRNA 假说选取RNA 之间表达量的负相关方法,来构建四个期型的CeRNA 网络。
我们发现网络由microRNAs 及其靶向的mRNAs (或者lncRNAs)组成,具有双二分图结构(Two-level bipartitestructure)。比较LUAD 四期网络中microRNA 的组分,CeRNA 网络自然划分为两类子网络:由全部期型中一组不变的microRNA 构成的共有的内源性竞争网络(The common competing endogenous networks, CCENs),以及不同期型特有的microRNA 构成的独有的内源性竞争网络(The unique competing endogenous networks, UCENs)。CeRNA 网络中mRNA 进行系统的基因本体(Gene ontology, GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)通路的富集分析,发现构建出的CeRNA 网络与癌症及其演化相关的通路密切相关,且随着LUAD 期型的恶化与癌症相关程度也更加紧密。另外,UCENs 中microRNA 相连接的mRNAs 相比于从CCENs 或者UCENs 中直接选取的mRNAs 有更高的富集效率。
另外主成分分析(PCA, principal component analysis)技术研究经过差异化表达分析的LUAD 样本数据,并结合Cox 等比例风险回归模型分析各主成分及其本征矢对病人生存情况的影响。通过对PCA 的本征矢进行层次聚类,我们发现一些在CeRNA 网络工作中被报道过的microRNA 能够显著影响LUAD 样本生存情况。Kaplan-Meier 生存曲线分析发现CCEN中六个microRNA 能影响LUAD 全部样本的生存情况,并且四个microRNA 能够影响LUAD 某一特定期型样本的生存情况。这十个microRNA 可以作为诊断和监视LUAD 的潜在标志物和预后手段。
PCA算法工作:
我们对于经过差异化表达分析的LUAD 一期、二期、三四期sample-RNA 表达数据,使用PCA 算法进行降维处理,减少组学数据的规模对于寻找疾病标志物研究的影响。Cox 等比例风险回归分析被用来确定PCA 重构数据对样本生存的影响,并对比性别或年龄指标,从而证明PCA算法在降低数据维度的同时,也在一定程度上保留了原始数据的生物信息。另外,通过对PCA 主成分对应的本征矢进行层次聚类分析,以及本征矢代表的RNA 与其在LUAD 样本中FC 值的相关性,来探究哪些RNA 在PCA 重构数据过程中对维持原始数据信息发挥重要作用。PCA算法及其Cox 等比例风险回归分析作为CeRNA 网络工作对于数据处理方式的补充,以及为寻找可能的生物标志物提供数据降维处理及生信挖掘。
CeRNA网络研究与PCA算法工作与的关系:
一方面,PCA 去除了生物体基因表达过程或者高通量组学数据采集过程中的噪声,很大程度上降低了原始sample-RNA 表达数据的维度,同时也保留数据一定的生物学意义,如对病人生存情况的影响显著高于诸如性别或年龄等指标。
另一方面,CeRNA 网络的方法通过选取microRNA 靶目标和筛选RNA 之间负相关连接,则更具有生物学意义,即很大程度上描述了机体内参与癌症演化的非编码RNA 之间的相互作用。
解读:
复杂系统理论是人们认识复杂世界及其变化规律的重要理论基础。随着大数据时代信息的爆炸增长,诸如交通人群大数据收集、生物组学信息采集等众多领域产生大量的实验数据,这就对数据处理和分析方法提出重大挑战。目前复杂网络的研究方法日趋成熟,越来越多的研究者也将其应用到更加广泛的学科领域范围。着眼于生物信息组学数据处理和非编码RNA相互调节的网络建模,重点研究了以下内容。
第一,基于疾病样本的临床数据和基因表达数据,构建非编码RNA之间相互作用的网络。
第二,将疾病划分为不同的演化阶段,研究疾病恶化过程中网络的相应变化。
第三,借助生物信息学的方法对网络的可靠性进行验证,并证明网络中的一些节点能够影响病人的生存情况。
第四,通过主成分分析技术对数据进行降维处理,结合风险回归分析、聚类分析等侧面验证上述结论。
亮点之一,系统地介绍了疾病相关的基因数据数理流程和网络构建的步骤,并通过对大量的网络节点的统计分析和生物信息相关的第三方数据库进行验证,这为进一步分析网络的性质和探索网络与疾病之间关系奠定基础。
亮点之二,作者发现构建的非编码RNA相互作用网络具有双二分图结构,并且网络的节点能够影响疾病恶化过程中病人的生存情况,因而通过构建网络的方法能够更好的寻找疾病的潜在药物靶点。
亮点之三,作者围绕寻找疾病的生物标志物为目标,通过采用基于生物基因组学数据的网络建模方法进行一系列的探索,并结合生物信息学方法进行结果证明,研究具有一定的可靠性,具有很强的实用价值。
工作及时地跟进新一代基因组学测序技术的最新发展,提出了一套数据处理和分析的框架,为复杂网络在基于生物数据建模方面提供解决思路,其方法框架也可以应用于更加广泛的生物数据分析和网络模型构建。这能够极大的扩展复杂网络的应用范围,以及复杂系统理论对其他学科领域的影响,加速交叉学科的快速发展,极具启发意义。
https://blog.sciencenet.cn/blog-3436387-1236987.html
上一篇:科研经历回顾--我的硕士阶段(自传性质)