博文
单原子到底有没有催化活性
|
编者按
“单原子催化”自从被大连化物所张涛老师等人在Nature Chemistry论文中提出以来,整个研究领域方兴未艾(具体发展历史,可以参见科学温故社往期推文:单原子催化的前世今生)。模型催化(如能够稳定单原子的Fe3O4(001)单晶研究)、粉末催化(CeO2、Fe2O3等氧化物以及MoS2和N掺杂石墨烯等二维材料作为衬底负载金属单原子)以及理论催化(催化反应机制的理解)都在单原子催化研究领域取得了一系列的成果。但是一个根本的问题: “单原子到底有没有催化活性”一直以来没有被重视(文献上报道的都是活性破纪录的各种单原子催化剂),直到2015年,来自美国西北大学的Peter C. Stair教授发表在Science的文章之后,算是第一次有人正视这个问题。我们知道,催化反应往往需要反应物分子共吸附于催化剂的活性位点(原子尺度催化动力学)。对于复杂的反应而言(如使用了碳数比较多的有机物,共吸附位阻太大),这个共吸附过程难以实现,因此目前选择的热催化反应绝大多数集中在CO氧化反应。即便是CO氧化反应,目前认可的反应机制也只是MvK机制(模型催化开山鼻祖Ertl教授工作知多少(二)),即吸附于单原子的CO与近邻的氧化物中的晶格氧反应后,形成CO2脱附之后,氧化物表面生成氧缺陷,随后O2在该缺陷上吸附解离,完成催化循环。然而Peter C. Stair教授却发现即使对于CO氧化反应,负载的Pt单原子也没有催化活性,这到底是因为什么呢?
四大法宝
目前单原子催化中研究的手段主要集中在高分辨电镜(HAADF)-STEM(用来确认单原子的存在,即电镜图中的亮点)、X-射线吸收谱 (XAS,确定催化剂的结构)、红外(IR,确定催化剂表面反应物以及中间产物的吸附)以及理论计算(DFT模拟反应机理,确定催化活性位以及反应机制)。这四种表征方法在张涛老师等人的论文中已经被多次应用,并且都能很好地解决问题。本篇推文中,我们着重介绍红外技术,因为该方法和催化剂的表面吸附物种相关,并可以表征反应活性。(红外方法可以参看:【课件】辛勤老师:催化剂活性中心/活性相的认知和调变)

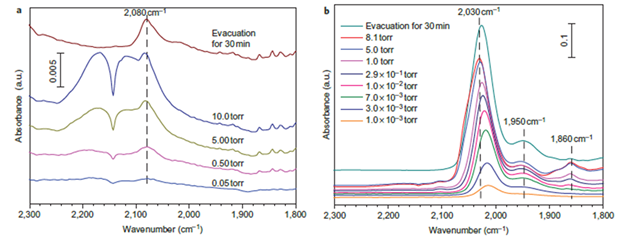
图1. 样品A( 0.17 wt% Pt/Fe3O4)和 样品B(2.50 wt% Pt/Fe3O4)CO红外吸附峰。
表1. CO氧化反应的活性对比
没有活性
该篇文章主要是借助于红外吸收光谱来研究负载型Pt催化剂催化反应的活性位以及碱金属(Na)对催化活性提升原因。
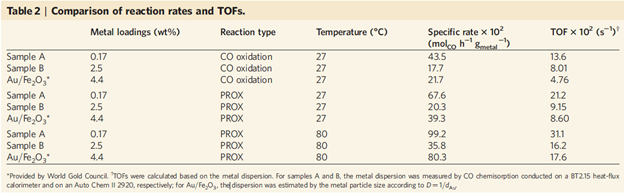
进一步地,作者进行了Pt催化剂表面CO的滴定实验:0.5 wt%的单原子Pt/ HZSM-5催化剂的CO红外吸收峰并没有随着O2的引入而消失,即使温度升高到100 oC,CO氧化反应依旧没有发生(图2c)。但是对于单原子和纳米粒子共存的2.6 wt%的Pt/ HZSM-5催化剂而言,随着O2的引入,Pt纳米粒子表面的线性吸附CO峰强度逐渐下降(图2d),在100 oC时完全消除,但是对应单原子催化剂表面的CO依旧存在。通过该对比实验可以发现,单原子Pt没有CO氧化活性,而Pt的纳米粒子在高温下可以发生CO氧化反应。同时从另外一个角度,单原子Pt表面的CO吸附键合相对于纳米粒子Pt而言更强。
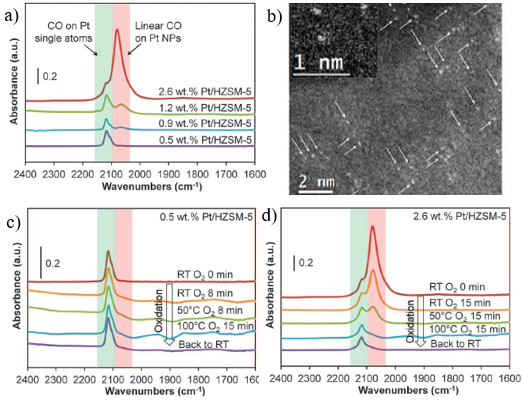
进一步地,作者将载体拓展到了一系列氧化物载体上(SiO2、TiO2、Al2O3以及ZrO2),制备出1wt% 负载型催化剂Pt催化剂。借助于CO的红外吸收光谱,发现氧化物负载的Pt催化剂基本上都有和Pt/ HZSM-5催化剂情况相似,都有类似的CO吸收峰。高分辨电镜确认了1 wt%Pt样品是由Pt纳米粒子和Pt单原子共同组成。当100 oC下引入O2之后,氧化物表面负载的1 wt%Pt催化剂中的Pt纳米粒子吸附的CO都可以被滴定掉,而其中的单原子的CO吸附峰的强度没有明显变化,再次说明单原子Pt没有催化活性。
再进一步,该作者将催化反应从简单的CO氧化拓展到了水汽变化反应(WGS)。同样发现,随着H2O的引入,单原子Pt表面吸附的CO依旧稳定存在,但是Pt纳米粒子上的CO 很快就被反应掉。伴随Na的加入,催化剂上明显看到CO的强度变弱,高分辨电镜确认了Na+可以显著提高Pt的分散度,因而相应地WGS催化活性也相应提升(图4)。
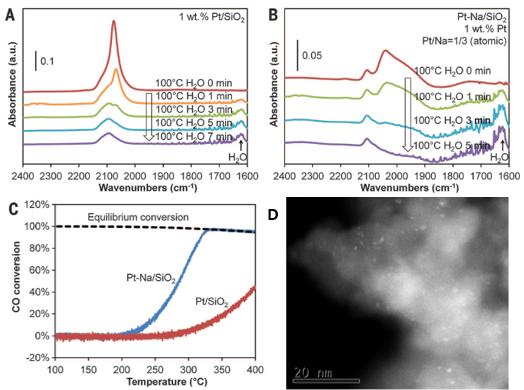
对比两篇文章我们可以发现,催化剂的组分基本相似,都是氧化物负载Pt。催化剂的结构也基本相似,都是低的担载量下Pt以单原子存在,高的担载量下,Pt单原子与Pt纳米粒子共存于催化剂表面。那为什么Nature Chemistry 文章中的催化剂有室温催化CO氧化活性,而Science文章中的催化剂活性就没有?对比催化剂的预处理条件发现,前者主要通过H2进行还原处理,后者主要通过O2进行处理。因此Science文章中报道的Pt处于更加氧化的状态,相应地,CO的吸附峰相较于前者发生蓝移,这与文章中红外报道一致。那这个预处理为什么会对催化活性又这么大的影响,导致不同的实验结论呢?

Science文章的表征手段更多聚焦于红外吸收谱的方法,通过CO吸收峰的位置来确认Pt的存在形式(Pt单原子以及Pt纳米粒子)。虽然也使用了高分辨电镜,但是分辨率一般。UCSB的Phillip Christopher教授课题组通过更可控的制备方法,制备出了TiO2负载的不同担载量的Pt催化剂,并借助于高分辨电镜以及红外对催化剂的氧化以及还原预处理过程进行了更加深入的研究:

对于240 oC的H2还原后的0.05 wt%Pt/TiO2催化剂而言,红外以及高分辨电镜(图5)确认了氧化物表面的Pt以单原子的形式存在,这个和Nature Chemistry报道的一致。而对于1 wt%Pt/TiO2而言,红外峰主要呈现出两个吸收峰的特征,并在单原子的位置同样存在一个小峰(注意两个图的红外吸收峰的纵坐标刻度不同),说明表面共存Pt单原子(极少量)、Pt的小团簇以及Pt的纳米粒子。
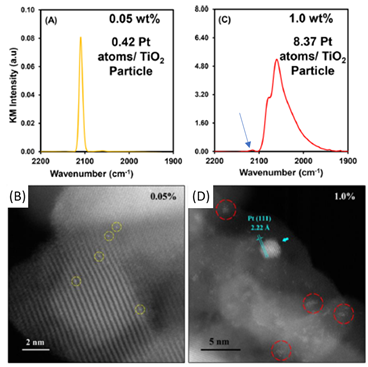
进一步地,作者研究了氧化处理对样品表面的CO吸附峰的影响(图6)。对于0.05 wt% Pt/TiO2的单原子样品,氧化之后整体的吸附峰的强度几乎降为0(注意图A中的纵坐标单位,其强度应该是相比于氧化处理之前降低了4个数量级,本来单原子吸附强度就很弱。单原子样品氧化之后,基本上没有什么CO吸附)。而1.0 wt% Pt/TiO2的纳米粒子同样氧化处理后,整个峰都往高场移动,开始还原状态下的很弱的2100 cm-1的吸附峰(源于单原子)的峰强明显增强,但是该峰的出现并不是代表氧化后单原子Pt的形成,而是由于氧化预处理导致的Pt纳米粒子带正电,整个CO吸附峰蓝移导致的。图6B图中的氧化后的1.0 wt% Pt/TiO2CO吸附峰和图2a中1.2 wt% Pt/HZSM-5的峰形基本一致。因而,单纯地通过峰的区间(2100 cm-1)来确定单原子的存在而相应忽视气氛处理,可能会导致催化剂结构归属的偏差。
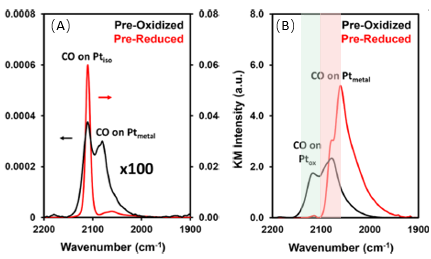
作者进一步指出,实际上当Pt的担载量提升到0.15 wt%时, 实际上无论从TEM或者是红外光谱(图7(A-B)),还原之后的样品都已经确定了表面已经是单原子和团簇共存的状态。相应地,也可以推测Science报道的负载的0. 5 wt %Pt的催化剂应该也是Pt的团簇和单原子共存,其氧化之后的样品的红外峰型基本上也是应该和图4类似,即2110 cm-1位置出现峰。同时,我们也可以推测,可能由于Science文章中的红外分辨率稍微差一些,这两个峰不一定能够完全分辨,因而很有可能出现的峰以2110 cm-1为主(图2a中的0. 5 wt %Pt/HZSM-5红外吸收峰有严重拖尾现象)。但是这个峰对应的应该不是Pt的单原子,而是氧化后的Pt的纳米粒子(因此文章中的电镜应该是有一定问题的,也就是看到的小亮点实际上不是单原子Pt,很有可能来自于其他污染,文章可能只关注与单原子了,看到纳米颗粒就直接草率的归结为电子束轰击的)。同样的,氧化状态的纳米粒子的活性是很低的,只有到了230℃下,高波数的CO吸附峰才部分反应掉(图7C)。从这篇文章中,我们有理由相信,Science文章中通过对分子筛或者氧化负载的1.0 wt% Pt上位于2100 cm-1处的CO吸附峰推测其来源于单原子上吸附的CO的结论可能是有一定偏差的;相应地,通过CO滴定实验说明Pt单原子的催化活性很差的结论出现了进一步的偏差的,催化剂的活性低应当来源于Pt的氧化物,而非单原子Pt。

图7. 0.15 wt% Pt/TiO2 催化剂的(A) CO红外吸收谱以及(B) 电镜图; (C)1 wt% Pt/TiO2 催化剂的CO红外滴定活性图。
个人思考
Wang, H. et al. Surpassing the single-atom catalytic activity limit through paired Pt-O-Pt ensemble built from isolated Pt1 atoms. Nat Commun 10, 3808 (2019).
https://blog.sciencenet.cn/blog-3438599-1254142.html
上一篇:李灿院士课题组进展梳理:光催化剂的晶面效应理解如何从现象上升到科学
下一篇:氧缺陷的多样性