博文
《光学学报》综述:多色单分子定位超分辨显微成像术
|
《光学学报》2017年第3期上推出“超分辨成像”专题。笔者根据自身研究经历,写了一篇多色单分子定位超分辨显微成像术综述,目前刚刚发表,分享给大家,请多多指点。
摘要 多色成像作为超分辨成像技术的重要延伸,极大地增强了人们研究亚细胞结构定位与交互关系的能力,从而有助于研究者深入理解细胞内复杂的生命现象与过程。单分子定位超分辨显微成像术由于其工作原理的特殊性,已诞生了激发依赖的、激活依赖的、分光依赖的等数种有特点的多色成像方法。本文介绍了六种主要的多色单分子定位超分辨成像技术,从分色能力、光谱窜扰、采集效率等角度分析了各方法的优缺点,并讨论了与多色成像相关的细胞固定方法,以期帮助研究人员根据自身实验需求选择合适可靠的多色成像手段探讨相应的科学问题。
1 引 言
远场光学显微镜因具备非侵入性、实时、可视放大观察样品等特点,自出现以来便成为细胞及微生物学研究中无可替代的常规技术手段。然而早在一百多年前德国物理学家Abbe指出由于光的波动性和衍射效应,使得远场成像系统横向分辨率存在约200 nm的极限,导致人们无法有效观测纳米尺度的生命现象。这一限制终于在21世纪初被打破,诞生了两大类超分辨荧光显微成像技术:1)基于激发光源改造(也称点扩散函数工程,point-spread-function engineering,PSF engineering)技术的受激发射损耗显微术(stimulated emission depletion microscopy, STED)[1-3]和结构光照明显微术(structured illumination microscopy, SIM)[4,5];2)基于单分子定位技术的光激活定位显微术(photo-activation localization microscopy, PALM)[6-8]和随机光学重建显微术(stochastic optical reconstruction microscopy, STORM)[9-12]。这些超分辨成像技术使得远场成像横向分辨率轻松达到了百纳米以下,进而大大丰富了显微镜对细胞微纳结构与功能的研究[13-18]。
基于单分子定位技术的超分辨显微成像(简称单分子定位显微术,single molecule localization microscopy, SMLM)通过“计算”实现,横向分辨率目前已达10 nm左右[19]。具体工作原理是:基于全内反射照明光路,用激活光和激发光控制成像区每次仅有少量、随机、离散的单个荧光分子发光,再通过高斯拟合分析单个荧光分子点扩展函数中心实现高精度的空间定位,最后将图片进行叠加形成一幅超分辨图像[20,21]。如利用荧光探针标记细胞内不同的目标物进行多色超分辨成像,不仅可以获得单个目标物的超分辨信息,还可以精确分析不同目标物的空间结构与相互关系,极大地加强人们对细胞内复杂的生命现象与过程的探索能力。目前,SMLM由于其工作方式的特殊性,已产生了数个不同原理的多色成像方法,并广泛地应用到生命科学各个研究领域中。其中代表性的工作是2013年哈佛大学Zhuang研究组采用双色STORM揭示神经细胞轴突部分微丝-帽蛋白复合物与血影蛋白交替构成间距为180~190 nm的周期性结构,这是传统研究手段无法观察到的[22]。本文将介绍几种主要的多色SMLM,依次阐述各种方法的优缺点,同时探讨与多色相关的样品固定处理方法,期望为不同实验需求的研究人员提供选择依据。
2 多色SMLM发展现状
多色SMLM按照采集方式主要可分为两大类,即依次采集(sequential imaging)和同步采集(simultaneous imaging)方式。目前存在以下几种主要的多色SMLM。
2.1 基于不同激发光的多色SMLM
依靠不同的激发光让不同染料分子发射出不同颜色的荧光是进行多色成像最常规的思路,已在PALM[23,24]与STORM[22, 25-27]中获得了较好的应用。尤其对于STORM,目前与商业有机荧光探针匹配的激光器波段主要包括488 nm,561 nm,647 nm和750 nm等,因此理论上可实现四色成像。该方法的优点在于工作原理简单,实施方便,不同颜色间窜扰较小。但缺点也比较明显,首先其属于依次采集方式,不同颜色间数据获取存在一定的时间间隔,则样品位置在数据采集过程难免发生漂移,因而颜色叠加时需进行校正(alignment)才能得到准确的相互关系。该问题目前可通过在样品中添加少量具有宽发射带宽的荧光微球解决[26]。其次,众所周知具有较强闪烁性(blinking)的荧光探针对于SMLM技术的实现至关重要[28],然而并不是每个激发波段对应的荧光探针都具有优良的闪烁能力。已有的闪烁性能较好的探针都集中在红外激发波段如Cy5,Alexa 647等,绿光和蓝光还没有对应闪烁性较好的染料,从而会严重影响组合成像分辨率,这也是该多色方法目前最大的问题。
2.2 基于不同激活光的多色SMLM
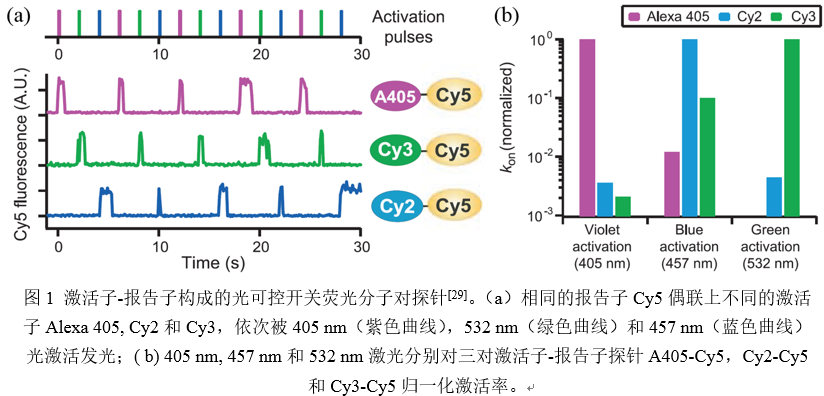
2006年,Zhuang研究组利用Cy3-Cy5(activator-receptor,激活子-报告子)组成的可控开关荧光分子对实现了单色STORM[9]。2007年,Zhuang组通过设计不同的荧光分子对组合,发展出基于不同激活光的多色STORM[29]。如图1所示,同一个报告子Cy5分别连接上不同的激活子如Alexa 405, Cy3和Cy2,经紫光,绿光和蓝光的激活后可发射出相同的红色荧光。这些荧光分子对通过连接二抗标记样品,依次切换上述不同波段的激活光,再由相同的激发光探测同一个报告子Cy5发出的荧光,从而实现多色成像。该方法特点是:1)可以自主选择闪烁能力最好的红外区报告子染料(如Cy5,Alexa 647等)进行单分子定位成像,有利于在每个激活通道获得高分辨率数据;2)其属于同步采集方式,即一次数据采集过程中获得所有颜色通道的数据,不同颜色间位置基本无位移,不需专门校正;3)把基于激发光和激活光多色成像联合使用,理论上可实现高达九色成像,目前已实现六色[30]和七色[31]成像。该方法缺点也比较明显,就是激发光本身会非特异性地激活不同颜色通道,导致较高的窜扰率(10~20%)[30],造成实验假象,尤其是待成像的两个目标物密度差异较大时,高密度目标物信号可能淹没低密度目标物[32]。
2.3 基于荧光淬灭技术的多色SMLM
2014年7月Tam等人发文报道了一种基于荧光淬灭(quenching)技术的多色STORM[33]。该方法原理简单,即分批次免疫荧光标记不同目标蛋白并采集数据,从而实现多色成像。整个实验过程需借助还原剂硼氢化钠淬灭上一轮目标蛋白二抗上的染料分子以避免窜扰,具体流程为:免疫标记目标蛋白1—>数据采集—>硼氢化钠淬灭—>免疫标记目标蛋白2—>数据采集—>硼氢化钠淬灭—>免疫标记目标蛋白3—>数据采集…。该方法属于依次采集,但只需一个激发光源和一个激活光源即可实现多色成像,颜色数只跟一抗种属(mouse,rat,rabbit,chicken等)数有关。此方法最大优点是不同目标蛋白二抗连接的染料分子是同一种,且选自闪烁能力最好的红外激发波段如Alexa647,因而保证了每个通道都具有较好的成像分辨率(优于激发依赖的多色成像),且由于淬灭技术导致几乎无窜扰(优于激活依赖的多色成像)。但是,不难看出整个实验过程非常的费时费力,本质上属于更加极端地采用了以时间换空间策略。此外,由于需要多次费时标记,对于不同颜色成像,需专门解决同一视野采集及位置校正问题。另外,由于是靠依次荧光淬灭实现多色成像,所以不适合PALM。
2.4 基于分光技术的多色SMLM
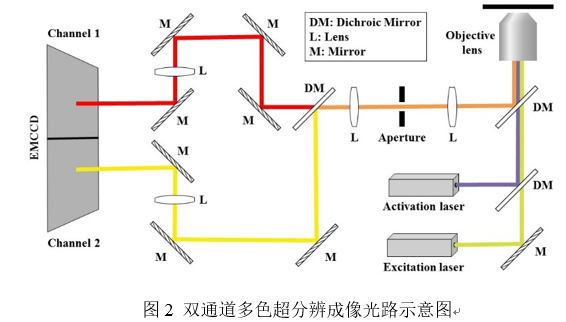
基于分光技术的多色成像是指采用不同发射谱的荧光探针标记样品,然后经同一个激发光辐照发射出的混合荧光被二向色镜分为透射和反射两路光,再分别被透镜聚焦到同一个CCD靶面左右两侧进行成像(图2)。数据采集后再利用比率法对每个荧光点进行颜色分类[34,35],最终获得双色图像,该方法已被应用于STORM[36,37],PALM[38]和STED[39]等多色成像。该方法主要优点是:1)其属于同步采集,只需要一个激发光源,一次数据采集过程同时获得两个颜色通道的数据,既节省实验时间,图片叠加也不需进行额外位置校正;2)可以选择闪烁能力较好的红外激发染料(如Alexa 647, CF680, Alexa 700等)标记样品,从而获得高分辨率数据。但该方法缺点是两个颜色间窜扰明显,尤其是待成像物密度较高的情况下更为糟糕。造成窜扰的原因主要有两方面:1)由于是一个激发光源,为了保证激发效率,所选的两个荧光探针的吸收谱必然近似,则它们的发射谱必然发生交叠从而导致窜扰,这是靠二向色镜和发射滤色片无法消除的;2)为了对两个通道的图片进行正确叠加,光路中二向色镜截止波段的选择往往刻意把其中一个波段的荧光分给另一个通道,使得两个通道数据产生关联性,进而实现校正叠加,但也带来窜扰,甚至是数据的丢失。该分光光路可以自行搭建,也可购买成品,如英国Andor公司的OptoSplit II和美国Photometrics公司的DV2。
2.5 基于光谱技术的多色SMLM
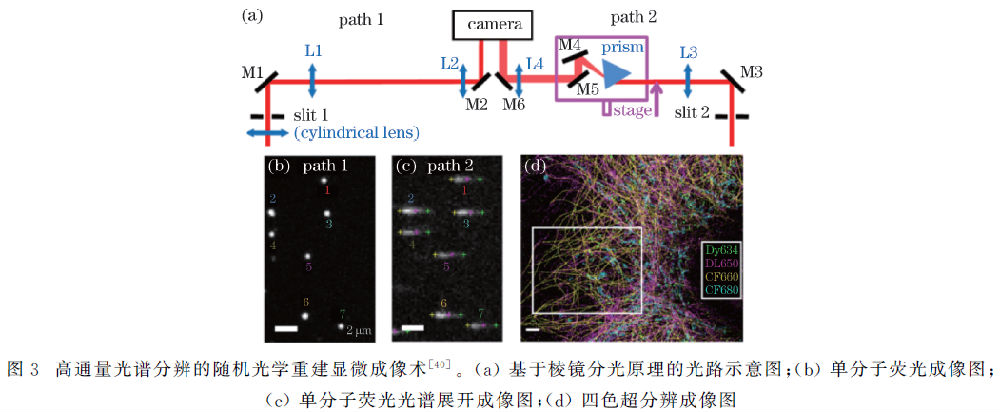
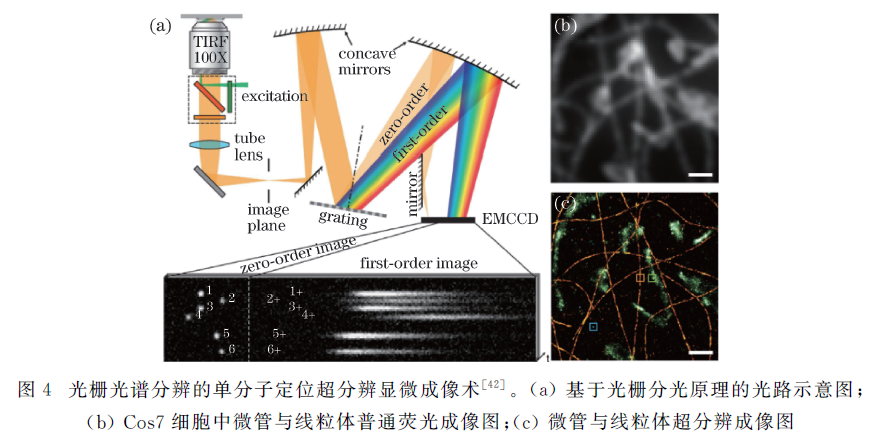
2015年8月加州大学伯克利分校Ke Xu研究组发文报道利用三棱镜分光原理,发展出一种名为光谱分辨的随机光学重建显微成像术(Spectrall Resolved STORM, SR-STORM)[40]。早在2013年,Xu利用双物镜成像系统提高荧光采集效率从而把STORM横向分辨率提高到10 nm左右[19]。基于该成像系统,他在其中一束光路中加入三棱镜(图3a),对荧光信号进行横向空间光谱展开(图3c),另一路进行正常的单分子空间定位成像(图3b)。最终计算出的超分辨图片中每个像点不仅具有空间信息,还拥有光谱信息,再通过单分子光谱比对与分类最终实现多色成像。文中为了降低光谱窜扰,筛选出Dy634、DL650、CF660和CF680四个荧光探针,在PtK2细胞中对过氧化物酶体、波形蛋白、微管和线粒体实现了四色同步超分辨成像(图3d)。该方法特点是:1)属于同步采集,只需一个激发光源和一个激活光源,可同时获得多达四色超分辨图像,且窜扰很低(< 2%);2)双物镜系统实现高通量荧光信号采集,获得光谱数据同时不影响空间定位分辨。问题是后期数据处理较为复杂。随后Hess研究组基于相似的光谱成像的方法,发现PALM成像过程中荧光蛋白的发射光谱可随时间发生变化[41]。2016年6月美国西北大学Zhang研究组发文报道利用光栅分光原理实现了光谱分辨的SMLM,并在Cos7细胞中对微管和线粒体进行了双色同步成像(图4)[42]。该方法主要特点是光谱展开较大,可获得较高的光谱分辨率。但由于是单物镜成像系统,光谱光路分流了本用于空间定位光路的荧光信号,影响空间分辨率;而且光谱光路只采用了1级衍射光,高级或者-1级没有利用,某种程度上降低了光谱分辨率。
不难理解,若希望光谱分辨率高,则光谱展开越大越好,这样光栅较三棱镜有明显的优势。但若展开过大,分到每个像素的荧光强度变低,导致光谱信噪比降低。当然,若展开不足,光谱分辨率太低,则无法进行光谱分析。此外,每个成像点进行光谱横向展开后,会占据较多的CCD靶面像素,荧光点随机闪烁过程中横向空间容易发生交叠,导致光谱数据失真,且原始图像点密度越高交叠概率越大。因此,不管是光栅还是棱镜分光技术,数据处理过程中必须把这些发生光谱交叠的数据点剔除,则导致对应的空间信息丢失,损失空间分辨率。此时存在一个三方博弈,即光谱信噪比、光谱分辨率与空间分辨率的取舍。对于细胞内结构的研究,各个目标蛋白位置和结构错综复杂,且密度较高,则棱镜法或许更为合适,可在保证足够的空间分辨率情况下实现多色同步成像。而若只是分析单分子荧光光谱动力学过程,且目标物密度较低,或许光栅法较合适。
2.6 基于点扩散函数工程技术的多色SMLM
2016年8月,诺贝尔奖获得者Moerner教授发文报道了一种基于点扩散函数工程(PSF engineering)技术的双色SMLM[43]。早在2009年,Moerner教授利用空间光调制器对单分子荧光进行点扩散函数双螺旋(double-helix point spread function)工程改造,使得荧光分子在不同的Z轴深度呈现不同的双螺旋调制信息,从而实现了三维SMLM[44]。随后,又基于分光技术进行点扩散函数双螺旋改造,实现了可定量分析的双色三维PALM[45]。2015年其利用四脚相位板对点扩散函数实现高精度超深三维调制[46]。2016年这篇文章合上述研究成果,采用单光路成像系统,设计出一种光谱依赖的四脚相位板,可Z轴深度依赖的调制不同颜色荧光的点扩散函数形状,进而实现双色三维SMLM(图5)。该方法概念新颖,属同步采集,但不依赖于分光技术,而是利用光学工程手段调制点扩散函数形状实现多色成像,颜色间窜扰较低。但也存在一定缺点,被调制过的点扩散函数占据了较多的CCD成像面积,限制了每帧图片的采集的分子数(否则信号易发生交叠),减低了采集效率,因此比较适合低密度目标物成像。

2.7 小结
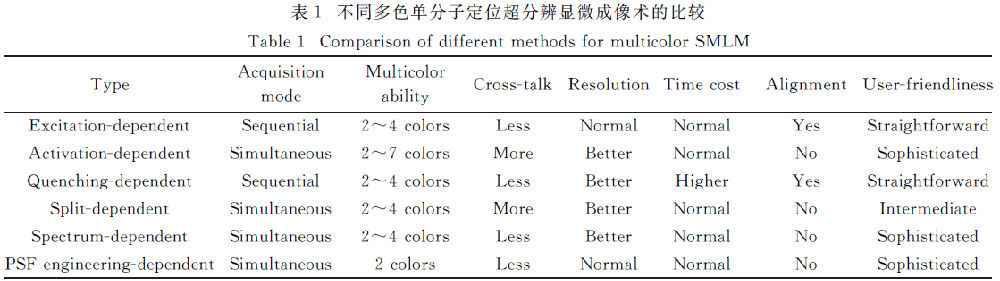
以上介绍了六种多色SMLM,原理各异,优缺点共存,具体比较如表1所示。论多色能力强,各通道成像效果均一,低窜扰等特点应选基于棱镜分光原理的SR-STORM,但是其数据处理较为复杂,不易上手;论原理简单、低窜扰当属基于不同激发光的多色SMLM,但不是每个通道都有适合超分辨成像的探针,稍有缺憾;论信号采集效率高及不需额外校正特点,当属基于分光技术的多色SMLM,但颜色窜扰固有存在。
3 多色成像中细胞固定问题
对于SMLM,工作原理要求被观察蛋白不能移动,因此一般需对细胞样品进行固定处理。PALM是利用内源表达的荧光蛋白成像,对固定要求不高。STORM是依靠免疫荧光标记技术,对样品固定要求比较严格,可以说适当的固定方法直接决定STORM图片质量[47,48]。目前细胞免疫染色常用的固定试剂有多聚甲醛溶液,戊二醛溶液,多聚甲醛+戊二醛混合液,冰甲醇等。以上固定试剂对细胞内不同的目标物会有不同的固定效果。多聚甲醛是最常用的固定试剂,渗透能力强,固定均匀,对目标蛋白本身结构影响不大,一般不影响一抗识别;但对膜的固定作用较差,不适合固定膜相关的结构,如内质网和膜关联蛋白。戊二醛是蛋白强固定剂,细微结构保持更好,对膜也有较好的固定作用,可真实的保存细胞的形态;但会影响目标蛋白空间构型,从而影响一抗识别,造成实验假象。冰甲醇通过蛋白沉淀固定样品,蛋白抗原性保存非常好,不影响抗体识别;但其会溶解膜结构,破坏细胞真实形态。对于多色STORM,本质是对多个目标蛋白进行成像,但各个蛋白结构特点不同,且对应抗体的性能和结合位点也不同,因此需根据具体实验目的探索合理的固定方法。研究经验表明,血影蛋白、帽蛋白、原肌球调节蛋白、带4.1蛋白等多色成像用多聚甲醛固定(~4%);微丝一般用戊二醛固定(0.1~2%);微管、线粒体、内质网等多色成像常用3%多聚甲醛+0.1%戊二醛混合液固定。但若进行微丝与血影蛋白多色成像,此时单纯的多聚甲醛(固定不住微丝)或者戊二醛(影响血影蛋白抗原性)可能都不合适,它们混合液可能比较合适,但需摸索出合适的浓度比。此外,戊二醛本身会导致细胞有较强的背景荧光,需用还原剂如硼氢化钠处理后方可实验,否则严重影响超分辨成像效果。
4 总结
本文介绍了六种多色SMLM的工作原理,比较了它们的优缺点,并探讨了多色成像中的细胞固定问题。多色成像是超分辨成像技术的重要补充,若想进一步推广多色SMLM在生物医学研究中的应用,笔者认为可在以下三个方面寻求突破:1)开发出闪烁能力强、发光效率高、颜色多样的有机荧光染料或荧光蛋白;2)开发出更适合探针闪烁的成像缓冲液;3)摸索出合理可靠的细胞固定试剂和方法。总之,技术本身无绝对孰优孰劣,关键根据自身研究需求选择合适的成像方法。
笔者曾于2015.12~2016.12在加州大学伯克利分校许可研究组从事STORM方面的研究,他是庄小威教授的博后,期间发表Science文章通过双色STORM揭示神经细胞轴突部分微丝-帽蛋白复合物与血影蛋白交替构成间距为180~190 nm的周期性结构(个人认为属于教科书级的工作)。笔者目前正在搭建STORM,欢迎大家一起合作探讨。
参考文献
[1] Hell S W, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy[J]. Optics Letters, 1994, 19(11): 780–782.
[2] Klar T A, Jakobs S, Dyba M, et al. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission[J]. Proceedings of the National Academy of Sciences, 2000, 97(15): 8206–8210.
[3] D’Este E, Kamin D, Göttfert F, et al. STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons[J]. Cell Reports, 2015, 10(8): 1246–1251.
[4] Gustafsson M G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy[J]. Journal of Microscopy, 2000, 198(2): 82–87.
[5] Gustafsson M G. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proceedings of the National Academy of Sciences, 2005, 102(37): 13081–13086.
[6] Betzig E, Patterson G H, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution[J]. Science, 2006, 313(5793): 1642–1645.
[7] Shtengel G, Galbraith J A, Galbraith C G, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proceedings of the National Academy of Sciences, 2009, 106(9): 3125–3130.
[8] Shroff H, White H, Betzig E. Photoactivated localization microscopy (PALM) of adhesion complexes[J]. Current Protocols in Cell Biology, 2008: 4-21.
[9] Rust M J, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)[J]. Nature Methods, 2006, 3(10): 793–796.
[10] Heilemann M, Van De Linde S, Schüttpelz M, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angewandte Chemie International Edition, 2008, 47(33): 6172–6176.
[11] Zhuang X. Nano-imaging with STORM[J]. Nature Photonics, 2009, 3(7): 365–367.
[12] Xu K, Shim S H, Zhuang X. Super-Resolution Imaging Through Stochastic Switching and Localization of Single Molecules: An Overview[M]. Far-field Optical Nanoscopy, Berlin: Springer, 2013, 27–64
[13] Yao Baoli, Lei Ming, Xue Bin, et al. Progress and applications of high-resolution and super-resolution optical imaging in space and biology[J]. Acta Photonica Sinica, 2011, 40 (11): 607–1618
姚保利, 雷 铭, 薛 彬, 等. 高分辨和超分辨光学成像技术在空间和生物中的应用[J]. 光子学报, 2011, 40 (11): 607–1618
[14] Xia Peng, Dou Zhen, Yao Xuebiao. Progress of super-resolution microscopy[J]. Chemistry of Life, 2015, 35(3): 460–437.
夏 鹏, 窦 震, 姚雪彪. 超高分辨率显微技术研究进展[J]. 生命的化学, 2015, 35(3): 460–437.
[15] Li D, Shao L, Chen B C, et al. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics[J]. Science, 2015, 349(6251): aab3500.
[16] Böhme M A, Beis C, Reddy-Alla S, et al. Active zone scaffolds differentially accumulate Unc13 isoforms to tune Ca2+ channel-vesicle coupling[J]. Nature Neuroscience, 2016, 19(10): 1311–1320.
[17] French J B, Jones S A, Deng H, et al. Spatial colocalization and functional link of purinosomes with mitochondria[J]. Science, 2016, 351(6274): 733–737.
[18] Huang F, Sirinakis G, Allgeyer E S, et al. Ultra-high resolution 3D imaging of whole cells. Cell, 2016, 166(4): 1028–1040.
[19] Xu K, Babcock H P, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton[J]. Nature Methods, 2012, 9(2): 185–188.
[20] Betzig E. Proposed method for molecular optical imaging[J]. Optics Letters, 1995, 20(3): 237–239.
[21] Dickson R M, Cubitt A B, Tsien R Y, et al. On/off blinking and switching behaviour of single molecules of green fluorescent protein[J]. Nature, 1997, 388(6640): 355–358.
[22] Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons[J]. Science, 2013, 339(6118): 452–456.
[23] Shroff H, Galbraith C G, Galbraith J A, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes[J]. Proceedings of the National Academy of Sciences, 2007, 104(51): 20308–20313.
[24] Subach F V, Patterson G H, Manley S, et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy[J]. Nature Methods, 2009, 6(2): 153–159.
[25] Dempsey G T, Vaughan J C, Chen K H, et al. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging[J]. Nature Methods, 2011, 8(12): 1027–1036.
[26] Jones S A, Shim S H, He J, et al. Fast, three-dimensional super-resolution imaging of live cells[J]. Nature Methods, 2011, 8(6): 499–505.
[27] Leterrier C, Potier J, Caillol G, et al. Nanoscale architecture of the axon initial segment reveals an organized and robust scaffold[J]. Cell Reports, 2015, 13(12): 2781–2793.
[28] Lippincott-Schwartz J, Patterson G H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging[J]. Trends in Cell Biology, 20019, 19(11): 555–565.
[29] Bates M, Huang B, Dempsey GT, et al. Multicolor super-resolution imaging with photo-switchable fluorescent probes[J]. Science, 2007, 317(5845):1749–1753
[30] Bates M, Dempsey G T, Chen K H, et al. Multicolor Super-Resolution Fluorescence Imaging via Multi-Parameter Fluorophore Detection[J]. ChemPhysChem, 2012, 13(1): 99–107.
[31] Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling[J]. Nature Methods, 2012, 9(7), 743–748.
[32] Dan A, Huang B, Bergan J, et al. Super resolution imaging of chemical synapses in the brain[J]. Neuron, 2010, 68(5): 843–856.
[33] Tam J, Cordier G A, Borbely J S, et al. (2014). Cross-talk-free multi-color STORM imaging using a single fluorophore. PLoS One, 2014, 9(7): e101772.
[34] Bossi M. Fölling J, Belov V N, et al. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species[J]. Nano Letters, 2008, 8(8): 2463–2468.
[35] Kim D, Curthoys N M, Parent M T, et al. Bleed-through correction for rendering and correlation analysis in multi-colour localization microscopy[J]. Journal of Optics, 2013, 15(9): 094011.
[36] Baddeley D, Crossman D, Rossberger S, et al. 4D super-resolution microscopy with conventional fluorophores and single wavelength excitation in optically thick cells and tissues[J]. PLoS One, 2011, 6(5): e20645.
[37] Lampe A, Haucke V, Sigrist S J, et al. Multi-colour direct STORM with red emitting carbocyanines[J]. Biology of the Cell, 2012, 104(4): 229–237.
[38] Gunewardene M S, Subach F V, Gould T J, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells[J]. Biophysical Journal, 2011, 101(6): 1522–1528.
[39] Testa I, Wurm C A, Medda R, et al. Multicolor fluorescence nanoscopy in fixed and living cells by exciting conventional fluorophores with a single wavelength[J]. Biophysical journal, 2010, 99(8): 2686–2694.
[40] Zhang Z, Kenny S J, Hauser M, et al. Ultrahigh-throughput single-molecule spectroscopy and spectrally resolved super-resolution microscopy[J]. Nature Methods, 2015, 12(10): 935–938.
[41] Mlodzianoski M J, Curthoys N M, Gunewardene M S, et al. Super-resolution imaging of molecular emission spectra and single molecule spectral fluctuations[J]. PLoS One, 2016, 11(3): e0147506.
[42] Dong B, Almassalha L, Urban B E, et al. Super-resolution spectroscopic microscopy via photon localization[J]. Nature Communications, 2016, 7: 12290
[43] Shechtman Y, Weiss L E, Backer A S, et al. (2016). Multicolour localization microscopy by point-spread-function engineering[J]. Nature Photonics, 2016, 10: 590–595.
[44] Pavani S R P, Thompson M A, Biteen J S, et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function[J]. Proceedings of the National Academy of Sciences, 2009, 106(9): 2995–2999.
[45] Gahlmann A, Ptacin J L, Grover G, et al. Quantitative multicolor subdiffraction imaging of bacterial protein ultrastructures in three dimensions[J]. Nano Letters, 2013, 13(3): 987–993.
[46] Shechtman Y, Weiss L E, Backer AS, et al. Precise Three-Dimensional Scan-Free Multiple-Particle Tracking over Large Axial Ranges with Tetrapod Point Spread Functions[J]. Nano Letters, 2015, 15(6): 4194–4199.
[47] Allen J R, Ross S T, Davidson M W. Sample preparation for single molecule localization microscopy[J]. Physical Chemistry Chemical Physics, 2013, 15(43): 18771–18783.
[48] Whelan D R, Bell T D. Image artifacts in single molecule localization microscopy: why optimization of sample preparation protocols matters[J]. Scientific Reports, 2015, 5: 7924
原文可以查找:潘雷霆, 胡芬, 张心正, 许京军. 多色单分子定位超分辨显微成像术的研究. 光学学报,2017, 37(3): 0318010
https://blog.sciencenet.cn/blog-382850-1060043.html
上一篇:超分辨光学成像引申讨论-时间信息和空间信息获取的博弈
下一篇:纸上谈兵—新冠肺炎发生与治疗