博文
应用贝叶斯谱系动力学解析我国TMV的时空迁移特征
|||
文献信息:
Gao, F., Liu, X., Du, Z., Hou, H., Wang, X., Wang, F., Yang, J., 2018. Bayesian phylodynamic analysis reveals the dispersal patterns of tobacco mosaic virus in China. Virology 528, 110-117. DOI: 10.1016/j.virol.2018.12.001
文献链接:
https://doi.org/10.1016/j.virol.2018.12.001
一、研究背景:
烟草花叶病毒(Tobacco mosaic virus, TMV)是烟草生产上最为常见并造成重要经济损失的植物病毒之一,也是第一个发现的植物病毒。该病毒作为一种模式病毒,在分子生物学、植物病理学等方面作了大量的研究,但对于其进化研究的报道较少。前人基于病毒-寄主共进化的假设估算该病毒的进化速率约2.20 × 10^−8 substitutions/site/year (Gibbs et al., 2010),而基于长期传代的实验进化研究发现TMV全基因组的平均进化速率为3.10 × 10^−4 sub/site/year (Kearney et al., 1999)。在TMV全基因组中,126K蛋白的进化速率较快,基于时间结构的序列数据(Time-structured sequence data)分析显示,其平均进化速率约为 7.90 × 10^−4 sub/site/year (95% CIs 3.90 × 10^−4–1.30 × 10^−3) (Pagan et al., 2010)。然而,关于该病毒的进化和群体动态的研究,尤其TMV我国不同烟草产区的迁移动态鲜有报道。本研究通过我国五个烟草主产区的10年采样并测序获得的TMV全基因组序列进行谱系动力研究,旨在为制定有效的TMV防控措施奠定理论依据。
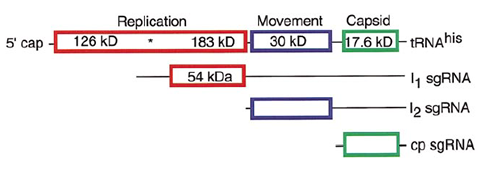
图1 烟草花叶病毒的基因组结构(Creager et al, 1999, Plant Cell 11, 301-308.)
二、方法流程:
1. TMV全基因组序列
(1)TMV中国分离物:56条,其中51条为本研究测得的TMV中国分离物,2009-2010年间采自12个省份的烟区,另外5条为我国早期报道的序列;
(2)其他国家TMV分离物:20条,包括法国、韩国、英国等国家,下载自GenBank数据库。
2. 重组分析和系统发育分析
采用两个重组方法分析,先应用SplitsTree 构建系统发育网络并计算成对同质性指数(pairwise homoplasy index, PHI查),随后应用RDP软件包中的7种方法,Bonferroni检验的p值设置0.01,至少四种方法均检测到显著(p< 1.0E-6)的重组事件视为有效。
系统发育分析应用ML法在IQ-TREE中分析,四个基因的核苷酸替代模型分别为GTR+Γ4, GTR+I, HKY+Γ4, K2P+I 模型,这些模型的准确性采用PhyloMad进行分析,结果详见文章的Fig. S1。
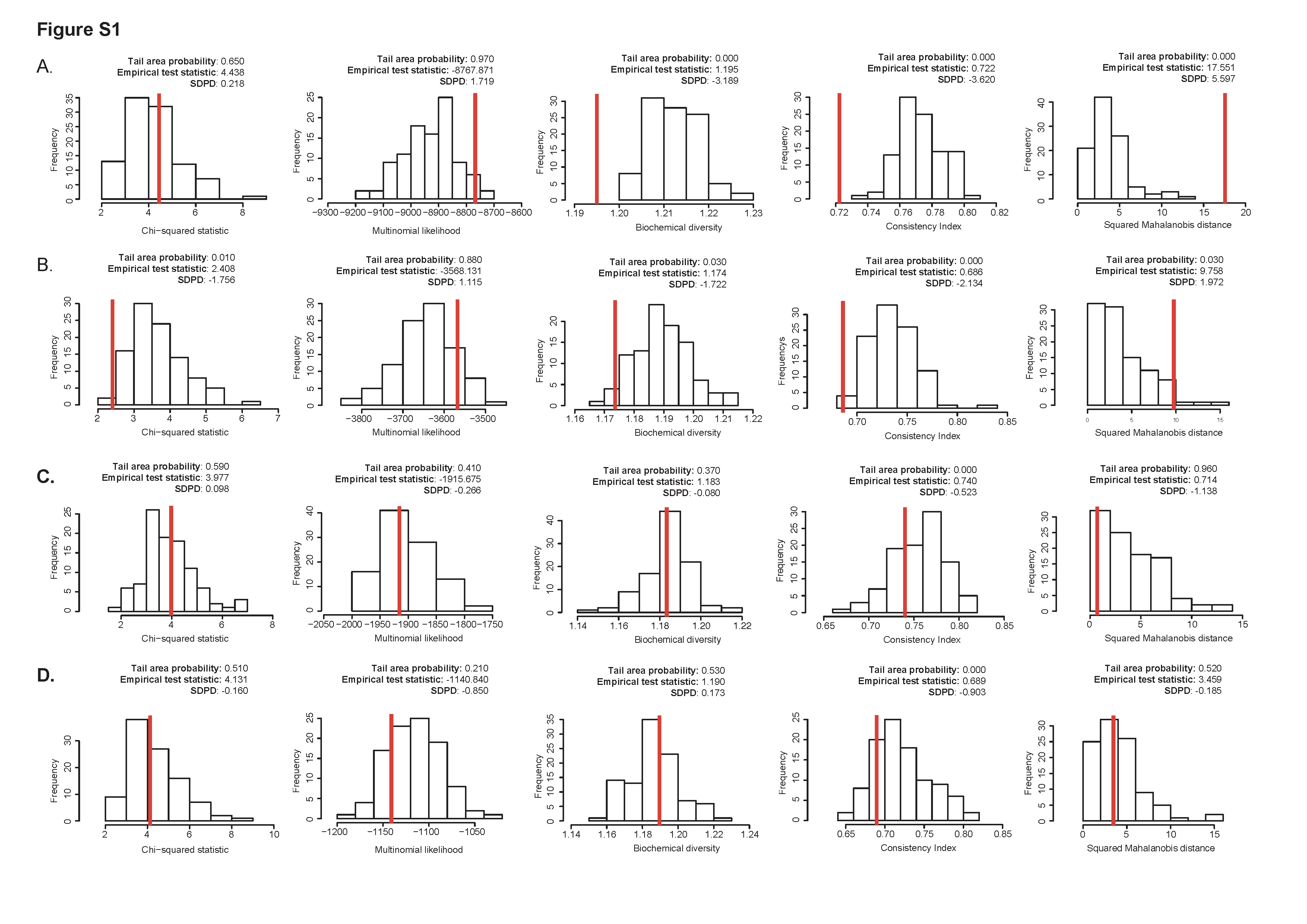
3. 时间信号检测
分别使用遗传距离和采样时间线性回归的方法,即可Root-to-tip(RTT)和日期随机化检验均未检测到足够的时间信号(Temporal signal),说明分析的TMV数据集为不具有时间结构(Temproal structure)的数据集,不用只用采样时间进行分子钟校准。
因此,根据前人报道的126K蛋白的进化速率进行分子钟校准,详见后述的主要难点及解决办法第一点。
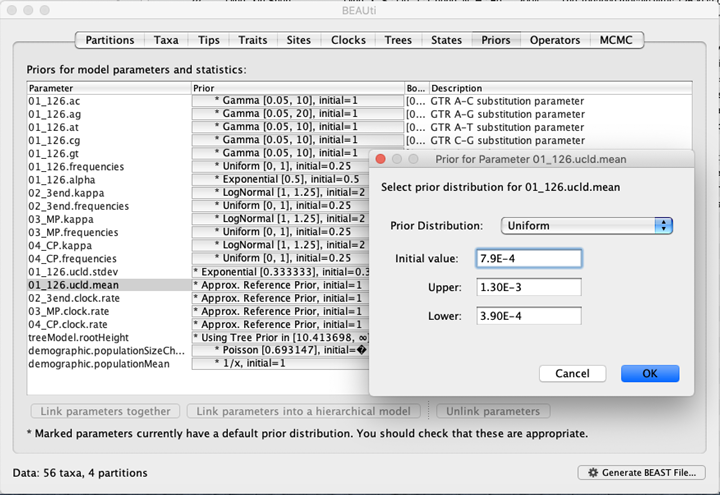
4. tMRCA和进化速率估算
在应用BEAST 分析TMV的tMRCA和进化速率前,,由于数据集为多基因联合分析,可将EBSP作为默认Tree Prior进行预分析(该模型可以推断有效群体大小随时间的变化),通过计算参数demographic.populationSizeChanges获得其平均值为2.4(95%HPD: 1-4),说明群体大小变化次数至少有2次及以上。因此,使用采用此Tree Piror适合分析的数据集,故而模型组合中只需要比较严格分子钟和宽松分子钟两组模型(Site 模型确定、Tree Prior模型确定)。分别配置后得到 Strict_EBSP.xml和 UCLN_EBSP.xml 通过PS/SS 法计算分别得到边际似然函数对数值为log marginal likelihood分别为-19,915和-19,706,表明宽松的分子钟模型更适合TMV数据集。其他参数详见文章中描述。
5. 群体分化检验
先定义不同烟草产区群体后,应用Arlequin软件通过计算两两群体间Fst值,根据计算的Fst 值确定群体间的分化度情况。
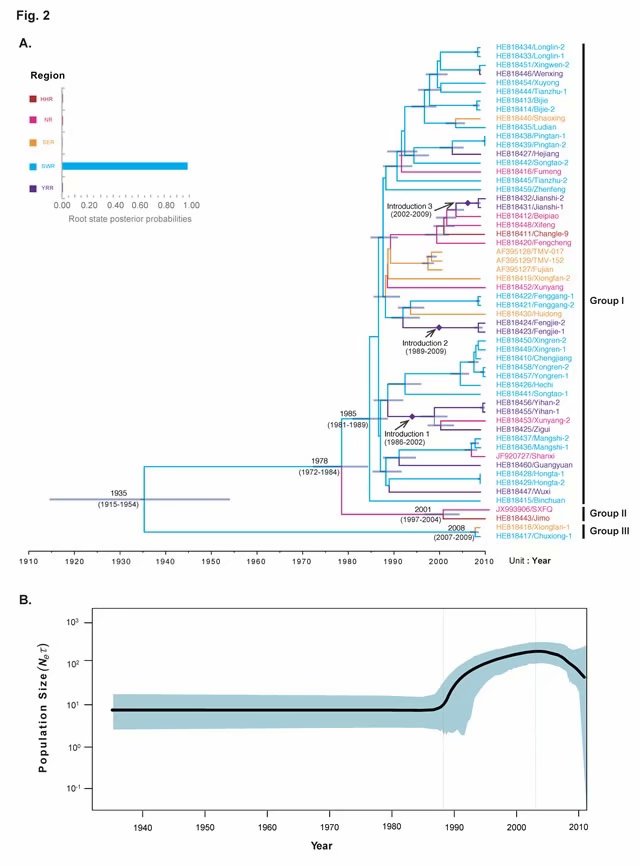
6. 谱系地理学和群体历史动态分析
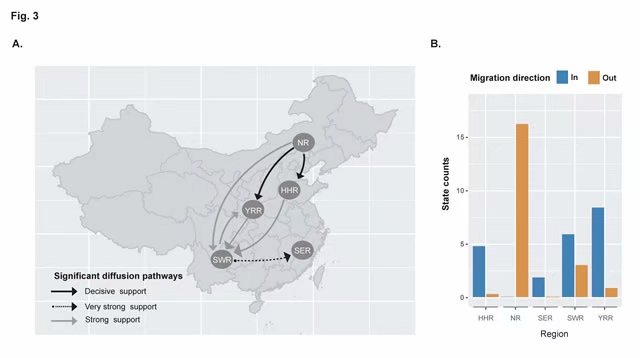
先将五个地区定义为Trait,应用贝叶斯随机搜索变量(Bayesian stochastic search variable selection, BSSVS)方法应用BEAST v1.84中重建 TMV不同产区群体间的迁移特征。通过贝叶斯因子法(Bayes factor, BF)在SpreaD3 v0.9.7计算获得BF值和平均后验值(Indicator)推断TMV两两产区间可能的迁移路径(至少BF>3且Indicator > 0.5时的迁移路径视为有效)。产区之间的迁移量通过BSSVS分析中的mean rate参数来衡量。在BEAUti v1.8.4 启用“不对称替换模型(asymmetric substitution model)”选项,其他参数配置参数详见日志《病毒的时空动态分析(BSSVS)简明图解》:http://user.qzone.qq.com/58001704/blog/1508718968。
三、研究结果:
研究发现烟草花叶病毒病毒(TMV)在20世纪初引入我国云南,随后扩散至北方,并在北方发生群体扩张。从新中国成立到现在,TMV总体上呈由北向南的流动趋势,这与我国的“北烟南移”政策紧密相关。该研究应用贝叶斯谱系动力学全面解析了我国TMV的时空扩散特征,可为该病毒的有效防控和精准测报奠定重要基础。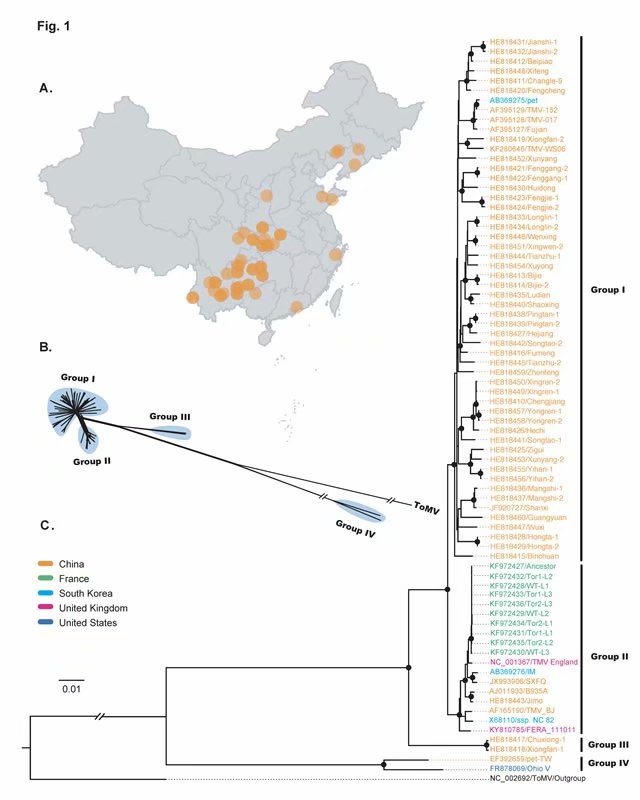
四、主要难点及解决办法:
(1)当数据集不具有时间信号不能直接用于分子钟校准时,如何解决?
当采样时间跨度大累积足够的遗传变异时,采样时间可以直接用于分子钟校准。这样的序列数据集被称为具有时间结构(Temporal structure)或时间信号(Temporal signal)的数据集。检测数据集是否具有时间结构或时间信号,可以通过 Root-to-tip(RTT)线条回归或日期随机化(Date-randomization test, DRT)进行判断。前者是一个较为粗犷的算法,在人畜患的RNA病毒的tMRCA和进化速率分析中较为常用,但其有个重要的前提,即基于严格分子钟假设,常用的工具有TempEst和TreeTime (https://treetime.biozentrum.unibas.ch)。而对通过DRTs 的数据集进行贝叶斯分析可以获得更为准确的tMRCA和进化速率,可以参阅 Guan et al, 2018, Virus Res., 256, 96-99 两篇文献。

图 x 时间信号检测 (Rieux & Balloux, 2016. Mol. Ecol. 25, 1911-1924)
而本研究的TMV数据集不论用RTT和DRTs均未找到足够的时间信号,因此解决的办法就是前人报道的126K蛋白的进化速率(平均值为7.90 × 10^−4 sub/site/year, 95% 置信区间为 3.90 × 10^−4–1.30 × 10^−3 sub/site/year)进化分子钟校准,该蛋白基因的进化速率也是推断自具有时间结构的数据集,详见 Pagan 等(2010) 原始文献。因此,在BEAUti设置Prior 时,校准点设置在126K蛋白对应的基因数据上,模型选择为Uniform,初始值设置为 7.90 E−4 sub/site/year ,下限为3.90 E−4,上限为1.30 E-3),其他3个基因允许有自己相对的速率。
参考描述:
A uniform prior of 3.90×10^−4–1.30×10^−3 substitutions/site/year was specified for the absolute substitution rate of the 126 K protein gene, based on a previous estimate (Pagan et al., 2010). The other three genes were allowed to have distinct relative rates.
(2)当不同地区样本量之间存在严重不平衡容易导致不准确的推断,如何解决?
本研究五大烟草种植区的TMV样本量存在不均衡情况,常见的解决办法有两种,一是基于结构溯祖的谱理地理学方法(如MASCOT、MTT等)可以有效解决样本量不平衡问题;二是本研究中应用的Bootstrap重采样法。
参考描述:
To investigate the impact of imbalanced sample sizes in our data set, we used a bootstrapping approach to standardize sample sizes and performed analyses of 10 replicate subsamples. For each bootstrap replicate, we randomly sampled 7 sequences with replacement from each tobacco-producing region. Each of these datasets was analysed using BEAST, as described above.
五、参考文献:
(1)Gibbs, A., Fargette, D., Garcia-Arenal, F., Gibbs, M., 2010. Time – the emerging dimension of plant virus studies. Journal of General Virology 91, 13-22.
(2)Duan, G., Zhan, F., Du, Z., Ho, S.Y.W., Gao, F., 2018. Europe was a hub for the global spread of potato virus S in the 19th century. Virology 525, 200-204.
(3)Guan, X., Yang, C., Fu, J., Du, Z., Ho, S.Y.W., Gao, F., 2018. Rapid evolutionary dynamics of pepper mild mottle virus. Virus Research 256, 96-99.
(4)Kearney, C.M., Thomson, M.J., Roland, K.E., 1999. Genome evolution of tobacco mosaic virus populations during long-term passaging in a diverse range of hosts. Archives of Virology 144, 1513-1526.
(5)Pagan, I., Firth, C., Holmes, E.C., 2010. Phylogenetic analysis reveals rapid evolutionary dynamics in the plant RNA virus genus tobamovirus. Journal of Molecular Evolution 71, 298-307.
https://blog.sciencenet.cn/blog-460481-1166970.html
上一篇:选择压力分析之EasyCodeML完整篇(By Raindy)
下一篇:多基因联合分析简明图解之PhyloSuite篇