博文
高通量识别核基因多信息位点在浅层次系统发育和谱系地理中的应用
|||
High-Throughput Identification of InformativeNuclear Loci for Shallow-Scale Phylogenetics and Phylogeography
高通量识别核基因多信息位点在浅层次系统发育和谱系地理中的应用
ALAN R. LEMMON1,* AND EMILY MORIARTY LEMMON2
原文:https://academic.oup.com/sysbio/article-lookup/doi/10.1093/sysbio/sys051
PDF: High-Throughput Identification of Informative Nuclear Loci for Shallow-Scale Phy.pdf
翻译:张仁杰
摘要
研究人员在研究浅层次系统发育和系谱地理问题的时候,主要的挑战之一就是为了他们感兴趣的问题而对变异度较高、信息量较大的核基因位点进行识别。之前关于基因位点识别的方法,往往需要对目标分类单元基因组库中未知的核基因位点进行大量的检测,并且还需对系统中其他未知功能的基因位点进行检测,或者对基因组中来源相近生物体模型的基因位点进行识别。在本文中,我们提出了一种快速并且节约成本的方法,对大量变异度较高、单次拷贝的核基因位点进行识别,以便之后可以在任一系统中进行下一代测序。我们选择在Illumina双端测序平台上,对来自拟蝗蛙属(Pseudacris)中三个库进行直系同源识别,并且在库中单次拷贝的基因位点水平和多重分类水平上评估序列的分异。我们也采用了PCR法对拟蝗蛙属间的这些基因和外群进行了测验,来确定位点在系谱地理学上的发展是否可以延伸到更深的系统发育层次。之前的测序,我们在电脑中分析了最相近关系的参照基因组(热带爪蟾),期待可以产生足够的基因位点数目和充分的覆盖度以满足我们实验设计的需求。使用RRL方法,我们可以:(1)识别超过100000个单次拷贝的细胞核基因位点,其中有我们所获得的分异的同一物种个体位点有6339个,异质个体位点904种(2)评估得出等位基因在个体内等位基因之间分异度为0.1%,可以代表两个不同进化分支的同种个体间分异度为1.1%、在不同物种之间为1.8%;(3)从PCR扩增确定:53%的基因位点可以在种内成功扩增,并且其中许多在属内也可以完成扩增,并且可以在更深的层次的系统发育水平进行扩增(16%)。在序列分异的基因组中,与之相似的线粒体基因位点相比,我们的研究目前已经可以高效识别核基因位点,可以用于系谱地理学研究中。特别地,我们评估了发现:在拟蝗蛙属基因组内约有7%的基因在种内的分异度>3%;这意味着在该基因组中大约50000个单次拷贝基因位点分异度>3%。此外,我们成功地在更深层次系统发育水平上扩增了大量的基因位点,这表明RRL方法可以用于系统发育和系谱地理研究中,并且是一种可以快速识别大信息量基因位点的方法。我们断定,在未来这一领域的研究中,这一方法可以将测序成本最小化并提高基因识别的效率。
关键词:未知核基因;IlluminaHiSeq;下一代测序:系统发育;系谱地理学;拟蝗蛙属;简化基因组文库。
为了获得最大效益,采用合适的方法识别大量高信息量的基因位点,是系谱地理学和浅层次系统发育研究中的一个重要瓶颈(Hare 2001)。例如,与核基因位点中潜在的高水平的系统发育信息相比,线粒体基因位点由于单一基因组可以在较短的时间进行融合这一优势,已经被广泛应用 (Neigel and Avise 1986;Hudson 1991)。此外,线粒体基因可以自由进行重组并且有着更小的群体有效性。越来越多的研究人员已经将核基因位点应用在系统发育和谱系地理的相关研究中,以避免单一依靠线粒体基因识别所产生的相关问题(e.g., selective sweeps, lackof clonality, nonneutrality, nonconstant mutation rates, and indirect selectioncaused by inherited symbionts: reviewed by Hurst and Jiggins 2005; Meiklejohnet al. 2007; Zink and Barrowclough 2008; Avise 2009; Brito and Edwards 2009;Galtier et al. 2009)。
许多工作者用于识别核基因位点的方法通常包括:(1)借用在其他系统中已被成功扩增的基因位点(e.g., Spinks et al. 2010;Fijarczyk et al. 2011);(2)从最相近基因组或者一套基因源中检测新的基因位点 (e.g., Townsend et al. 2008; Thompsonet al. 2008);或者(3)通过从一个基因库里进行序列克隆来识别未知核基因位点(e.g., Jennings and Edwards2005)。通常,我们通过Sanger测序,以及进行PCR扩增来获取序列数据,并且目标基因位点的选择,是基于是否利于扩增而不是基于基因位点的信息量。通过以上的方法,被选中的基因位点必须保留启动区域以便进行扩增,在序列变异之前可以得到评估等特点。实际上,这个需求可能偏向对低水平变异的基因位点的使用,并且因此影响我们通过核基因位点来解决浅层分异的效果(Hare 2001; Zhang and Hewitt2003; Avise 2009; Brito and Edwards 2009)。(叙述以往方法的不足)
本文,我们使用了RRL方法(reduced-representationlibrary)与NGS方法相结合来发现直系同源,以及种内和种间高度分异序列的单次拷贝基因位点。RRL法是通过用一到多种的限制酶基因组DNA切片,在凝胶上消化吸收样品,然后在多个个体间选择相同尺度大小的片段。例如,RRL测序和相关方法已经 (e.g., restriction-site associated DNA(RAD) sequencing, Baird et al. 2008)解决了复杂遗传图谱中单一核苷酸的多态性问题并且在群体遗传学中已经有所应用(e.g., Wiedmann et al. 2008; Kerstens etal. 2009; Emerson et al. 2010)。原则上,RRL法允许研究人员选择一个基因组的子集,这些子集包括不同个体间相似度极高的基因区域;这个子集同时包括编码区和非编码区(Altshuler et al. 2000)。这个方法缓解了对保留启动区域的部分限制,比如必须使用短限制位点(e.g., 6 bp)而不是长启动位点(e.g., >18 bp)必须被保留以产生数据,从而用来进行初步的序列分异评估。为了得到之后的PCR扩增序列,我们在Illumina 2000测序平台上使用双端测序(paired-end)来识别成对的启动区域。通过提供一种快速的方法,来发掘细胞核基因组潜在信息基因位点。这个方法规避了一个在系统发育学和系谱地理学中的实质性问题。高处理量双端测序序列的利用可以帮助避免等位基因校正的问题(Avise 2009, see Discussionsection)。除了发展这一新的方法外,我们在本文中还增加了两个问题:(1)在不同分类尺度下我们所获取的基因位点会产生多大的变异度?(2)这一方法怎样在更深层次的系统发育尺度下发挥作用,例如在种间和属间?
理想地,我们想要对选取不同的基因位点来对进化速率进行评估。然而,在浅层次的系统发育水平评估进化速率十分困难,因为我们需要直接测量的数量(序列分异)是基因进化率和结合时间的一个函数(Hudson 1991)。这里,我们简要描述了一些序列分异基因位点最新的识别方法,然而在短时间尺度下,序列的分异可能不是进化率最完美的的预测指标。实际上,我们认为:虽然细胞核基因组的平均进化率实际上慢于在线粒体基因组中的进化率,但细胞核基因组有更广泛的分布率,我们认为它实际可能包含大量对系谱地理学上有效的基因位点。我们的目的之一就是评估在几种时间尺度下单次拷贝的细胞核基因序列变异的分布。我们保留了这些基因位点的分类,以便今后对其联合或然性以及未来关于进化率的研究。

方法
概述
本研究最初是想通过利用信息量丰富的细胞核基因位点来研究北美拟蝗蛙属谱系地理的历史(见图1)。该种的两个主要分支被认为在大约2600万年前享有共同的祖先(Ma; Lemmon et al. 2007; Lemmon andLemmon 2008)。我们也希望通过利用多信息量的基因位点,来更充分地研究拟蝗蛙属的系统发育问题(图1; Lemmon et al. 2007),其最初的分节估计在27Ma的时候(图1; Lemmon et al. 2007)。我们的实验材料来自P. feriarum每个分支的一个个体以及P. maculata中的一个个体,我们的目标是在这三个个体间获得大量的核基因位点,用已识别的的基因位点来评估序列变异,并且有计划的从这些基因位点中检测引物。为了发现这些基因位点,我们对大量长度约为600bp的基因片段进行测序。由于我们也希望在三个个体间获取同源基因,所以我们通过使用限制酶来分解基因组DNA,通过凝胶的方法来处理片段,并且从这三个个体中选择相同的大小范围。完成双端测序后可以获得每个序列为100 bp序列片段的数据。在对每个个体进行单次拷贝装配之后,在个体之间每个基因序列被连成一条直线并且自行设计引物。利用来自所有Pseudacris及其他几个外群的大型panel 对引物进行检测。
本次实验中,我们采用了三种特别的实验设计以减少在非模式生物有机体中使用一种新的分子技术所带来的困难:(1)在库的准备期间我们使用了几种不同的限制酶(2)对关联程度不同的多个个体进行测序(3)收集大量的测序数据。研究的目的之一是为了挖掘这些必要的元素,以致于我们可以将其推荐给研究人员使用RRL的方法对非模型的生物有机体中的核基因位点进行识别。因为我们需要在属(拟蝗蛙属)的水平上识别基因位点,但其中最近的有效的基因组分异时间较长(在非洲爪蟾属上的分异时间为~200 Ma; Pyron 2011),并且只采用一种限制酶很可能改变基因组的容量,因此我们使用8种不同的限制酶去预测跨越四个不同数量级的基因位点数量。由于我们希望确定在不同时间尺度下核基因位点间序列变异的分布,所以我们对两个个体使用了RRL方法:其中一个代表了物种不同分支,另一个代表了一个来自其他物种的个体,这两个分支的分异相隔9600万年(Lemmon et al. 2007)。最后,我们搜集了大量的序列数据(three Illumina HiSeq 2000lanes),以确保我们可以获得足够的覆盖率,产生大量的基因位点从而对杂合基因型进行准确识别。在讨论中,通过我们的结论指出:对于在其他非模型系统中挖掘大信息量基因位点最高效的方法。方法的简单工作流程在图2已经给出。
通过生物信息预测,选取限制酶
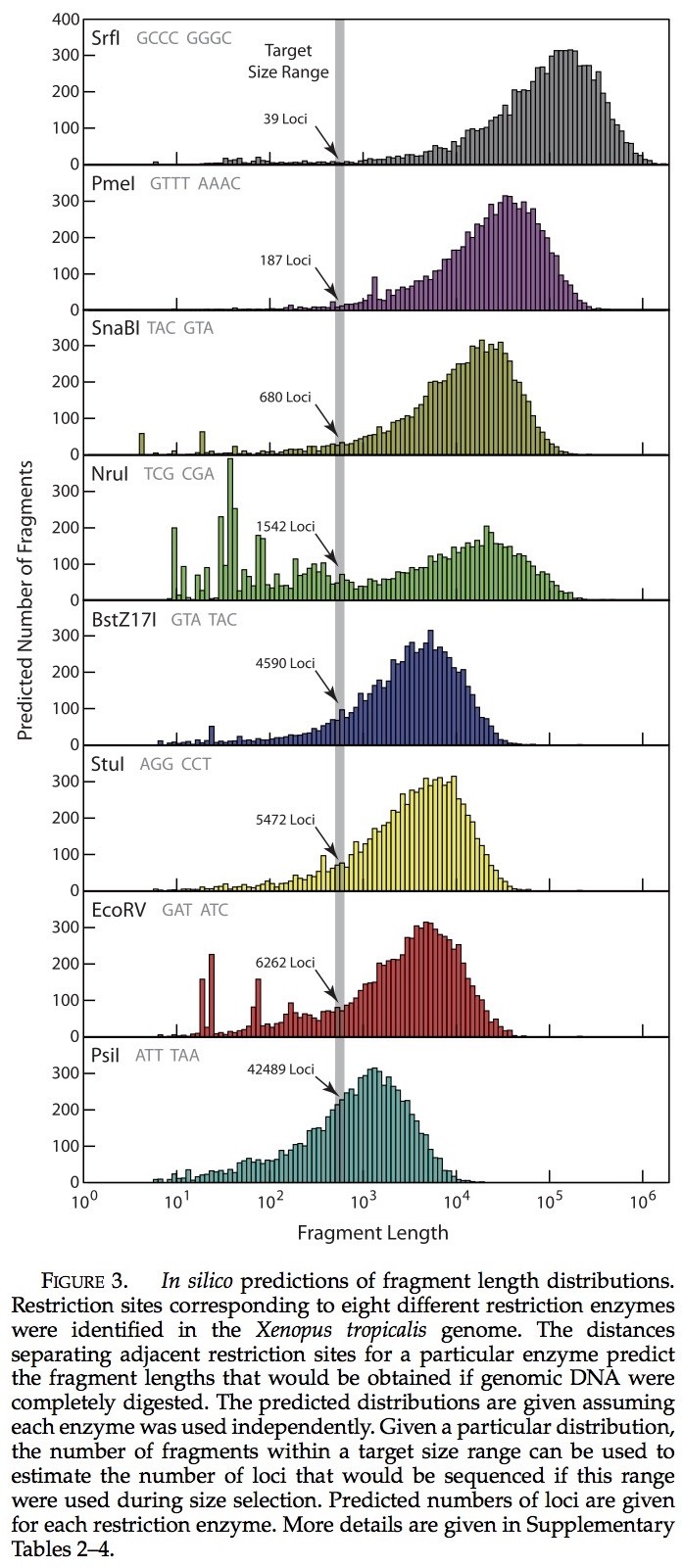
使用非洲爪蟾蜍基因组进行生物信息预测,将其结果作为选择限制酶的依据(Xentr4, v.4.1, August 2005, Joint GenomeInstitute ,http://genome.jgi psf. org/Xentr4/Xentr4.info.html)。在获得基因组序列之后,通过分别使用25个不同的2型限制酶我们评估了片段的长度分布,这种回文结构的限制酶其相应的限制位点在长度上超过了5 bp(补充表1)。对于每种限制酶,我们预测片段长度的分布可能源于对非洲爪蟾蜍基因组的100%消化(补充表1)。简单地说:(1)承载基因序列数据(2)在每个限制位点将序列分裂为更小的序列(3)计算所得子序列的长度。我们选择了8种限制酶(SrfI, PmeI, SnaBI, NruI, BstZ17I, StuI,EcoRV, and PsiI),它们可以代表所观察的分布范围(图3),但是当它们一起使用时,也可以在每个位点的碱基组成中产生许多变化。之后的要求是十分重要的,因为Illumina HiSeq测序软件需要在需要变化的最初几个碱基中有变异产生,以便识别准确。利用这些预测片段长度的分布,我们鉴别出一个大小范围:514–634 bp,当使用单一Illumina HiSeq 2000 测序平台在该长度范围下对resulting library进行测序的时候(补充表2-4),可能会产生完全覆盖的大量基因位点。由于每种酶的差异所产生的片段大小的分布不同,预计这8种酶的标记在所选择的大小范围内会产生不同数量的基因位点。通过使用Dryad (http://datadryad.org,doi:10.5061/dryad.vh151q1c),以上的方案进行生物信息预测实验是可行的。
DNA提取、数据库的准备、测序
将三种拟蝗蛙(二倍体)收集到ACUC protocol #0905,并且使用E.Z.N.A. Tissue DNA Kit将其DNA从肝脏组织中提取出来(OmegaBio-Tek, Norcross, GA, USA)。样品包括了Pseudacris maculata(ECM7278; Boone Co., MO, USA),一个来自内陆分支的P. feriarum (ECM7210; Lafayette Co., MS, USA),还有一个来自沿海的分支的P. feriarum (ECM7144; Prince Edward Co.,VA, USA)。全部三种样品的收集都在与其他物种关联很近的异域性地区,从而减少杂交配种测序时异种特性的等位基因。
每个DNA样品被分别放在8个不同的限制酶中进行分解(EcoRV,NruI, PsiI, StuI, BstZ17I, PmeI, SnaBI, and Srf1)。随后在自动化电泳系统中分解DNA (Bio-Rad),我们在电脑中预测(见之前的介绍)得到验证结果。如果相关经验性分布得到一个相似的模型,在电脑中模拟可以得到验证。来自三种拟蝗蛙样品的每个DNA要被每种酶分解两次。协议要求酶供应商遵循以下原则:2L限制酶,2gDNA,10L 10× NEB缓冲剂,1.0 L Bovine Serum Albumin(BSA),还有加入足量的H20使总反应的容量达到100L。样品在37◦C条件下培养2小时,然后将温度保持在4◦C。两种消化酶合并之后加入到个体中,用MinElute PCR纯化工具(QIAGEN)进行纯化,加入20L洗脱液进行洗脱,并且在4 ◦C的条件下储存。
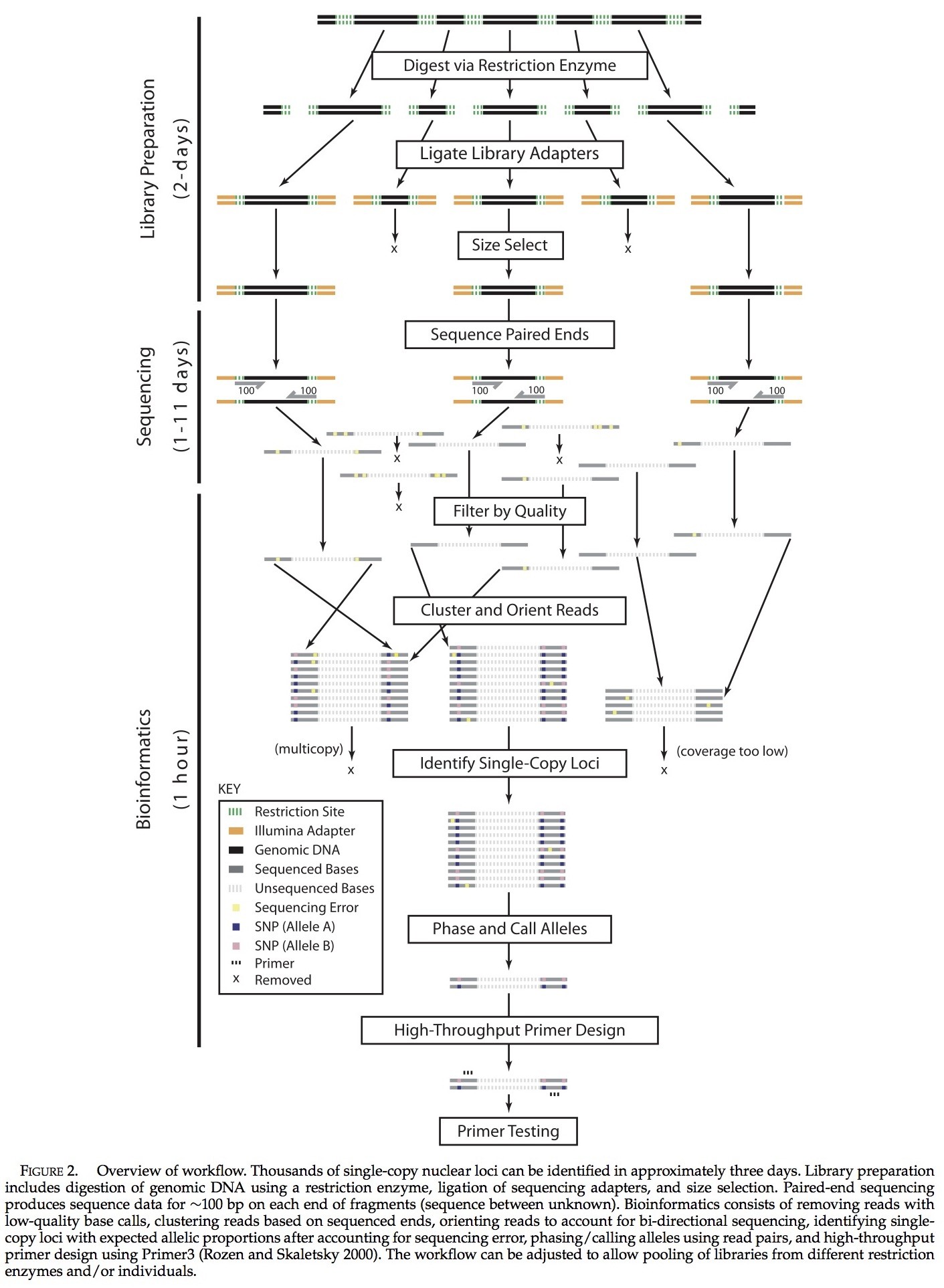
数据库的准备和测序在美国亨兹维尔市的生物技术研究院进行。在Qubit™荧光剂下对24个样品进行更广的范围和高敏感度的定量分析(每个个体分别使用8种酶)(Invitrogen, Carlsbad, CA, USA),并且将每个个体的酶在等摩尔比值下合成。Illumina双端测序库的准备在三个样品(个体)分别进行。通过在2% 1 × Tris-Acetate-EDTA (TAE)琼脂糖凝胶中进行双向电泳来对这三个库进行随后的个体选择(The three libraries were thenindividually size-selected by running samples for 105 min on a 2% 1 ×Tris-Acetate-EDTA (TAE) agarose gel),切割片段的范围从600–720 bp(该长度是由86bp的寡聚核苷酸组成),并且使用凝胶提取。这三个库在Agilent DNA 1000上进行分析以检测合适的范围,且集中并稀释到10-nM以便测序。用v2化学过程(每个库一条线)IlluminaHiSeq 2000测序系统中对100 bp的序列对进行测序。
生物信息预测
装配之前,测定的序列进行质量筛选并通过限制位点排序。质量筛选包括移除在第一个70bp质量分数少于20的包含一到多个相应片段全部序列对(<99%的准确率)。在质量筛选后,根据产生序列碎片的限制酶来对序列对分类。分类是可能的,因为这些特殊的限制酶产生的碎片有一半的限制位点,在每个序列的测序对的5’末端。。从更深层次的分析,被移除的序列使成对的5’末端不能与8个预期的限制位点序列相匹配。
在对第一个70bp的序列进行正向和反向读取的基础上,对序列对进行装配。我们采用了Illumina公司测定的序列有着非常低的读取错误率这一优点(<0.01%; Glenn 2011),以及充分的限制消化可以在序列中相同的每个位点产生一组片段(除了真正的多态性)。集群包括两步:(1)我们使用hashing法将序列对聚集起来(with akmer size of 70),这些序列对在每个序列的5’端都共有一个70 bp的序列。自从序列配对后,一对序列中的一个序列有错误,可能不会必然导致来自一个基因位点的序列被分成两丛,然而一个序列对中的每个序列都发生错误时将会导致序列分裂(但是可见下面的Basic Local Alignment SearchTool (BLAST)程序)。(2)成对测序使读取最大化,从两个方向的双链片段测序都可进行。最终,群集的序列呈直线排列,产生一个新的集合,因为对于区分来自真实多态性的序列错误,10是最小值,所以少于10个序列的群集将被移除(图2)。只有140bp的序列对被使用,因为初步分析认为全部200bp的每个序列对会导致SNP出现偏差。这种偏差源自序列变化是作为位点位置的一种函数而增加(与序列中的位置相一致,见Sequence Divergence的结论)。
每个集群的原始程序集评估证据显示这些序列取自不止一个基因位点。对于每个基因位点和每个位置,我们计算了来自每六个模型中观察到位点图示的可能性(两个单一基因位点和四个双基因位点模型):AA,AB,AA/AC, AA/BC,AB/AC,AB/CD,例如:AA/BC指出双基因位点模型的第一个基因位点包含一个等位基因,第二个基因位点包含两个附加的等位基因。图4表明每个模型的预期图示。
我们的计算结果认为,测序错误目前在0.01的水平上(相当于允许的最小phred质量分数,20,见上文),并且等位基因和特定的基因位点偏离在覆盖范围内消失。我们用这些可能性移除了一些分支,以使得最适模型中任一位点包含至少一个基因,正如在AIC 测试中所决定的那样(Akaike1974)。我们之后移除了分支,在特殊位点选择模型中,其片段无法叠加到三个等位基因以下(计算测序错误后,位点图示并非一致的多态性位点)。随机选取分支的一个子集通过肉眼进行评估,以验证这种方法是否产生了合理的结果。
BLAST是用于检验在等位基因的基因位点最终分支。更特殊的是,我们使用BLASTv2.2.23+(Camacho et al. 2009)对个体内和个体间的全部等位基因进行全部的比对(e值最大为0.001)。在个体内的比对允许有差异的等位基因组合到同一基因位点,然而个体间的对比采用建立同源性分析来进行。所有等位基因在BLAST配对中,个体中超过1个的额外的等位基因在随后的分析中被移除(包括在种间分析)。这种筛选减少了属于单次拷贝基因位点的等位基因。
同源基因呈线性排列:(1)在每个个体内;(2)在所有个体的成对组合间;(3) 使用默认参数通过Log-Expectation (MUSCLE; Edgar 2004),在全部三个个体内进行多重序列对比。我们采取两项措施,以确保准确估计在种群内和种群间的序列分歧。第一,我们移除了包含一个或多个间隙相应的排列一致的所有基因位点。第二,我们移除了在相应线性排列中第一个或最后一个3bp中,所有含序列分歧的两个等位基因的基因位点。这样做是因为初步分析表明,在引进接近末端的间隙对准时,MUSCLE有时可能失败,我们不想解释这些未校准位点的多态性。这两个严格的方法将会使得校准更加清晰准确。最终,随后我们将未修改的成对序列差异在每个个体内的等位基因间以及个体的基因位点中计算出来。所有线性估计的序列分歧>3%在视觉上可以被检测出,并且可以表明错误的证据(例如,由于较短的导致或聚物延伸)已被移除。采用Python脚本进行序列读取,序列聚集,拷贝数分析,等位基因识别,以及使用Dryad对等位基因进行移除是可行的(http://datadryad.org, doi:10.5061/dryad.vh151q1c)。
使用高通量方法将每个基因位点配对的引物序列选出。我们一开始就通过使用来自物种内和物种间共同序列对齐每个位点。单机版的Primer3 (Rozen and Skaletsky 2000)用于鉴别每个基因位点和分类等级上超过10000的引物对,要求每对包括一个引物,从每70 bp一边对齐(对应的片段结束)。引物包含来自多态性的未知碱基是不可用的(N或者其他不明确的IUPAC)。默认参数采用如下方式进行调整:PRIMER_MIN_SIZE=18,
PRIMER_OPT_SIZE=20, and PRIMER_MAX_SIZE=24.通过引物3对表中的10000个位点进行识别,我们选取了可以产生最长扩增子的引物对,通过选择在Primer3输出文件被列在第一位引物,将其纽带关系打破。
由于我们的目标是为了识别核基因位点,我们认为:最终的位点不是源自线粒体DNA。特殊的是,我们使用了BLAST(v2.2.23+; Camacho et al. 2009)将最终的等位基因序列与H. japonica线粒体基因组中的序列进行对比(Igawa et al. 2008),这是线粒体基因组中可用的最相近的一个物种(其与Pseudacris的分异时间在~44.7 Ma; Smith et al. 2007)。BLAST的默认参数可以使用(maxe-value = 10)。

在物种内测试候选基因
为了检测种内候选基因位点的利用,我们通过PCR,采用之前描述的引物将P. feriarum基因位点扩增至96个。基因位点在引物的熔融温度下被合并到群体内并且使用5◦C退火温度进行检测,该温度低于群体内引物最低熔融温度。群体内的退火温度有:45◦, 47◦, 48◦, 49◦, 和 51◦C。在这个研究过程期间,没有任何优化,我们在每个退火温度下对每个位点进行了检测。PCR在来自P. feriarum的两个分支中起到了代表性作用(ECM7145 and ECM7522; 补充表5)。,如果一个基因位点可以在这两个个体扩增并且单一条带出现在我们所预期的范围内,那么该基因位点被成功归类。这套成功的基因位点中,有20个在桑格测序中对每个位点间的序列分异进行了验证 (详情见下文PCR及测序反应;补充表6)。对两个个体中的这些位点进行了桑格测序——这两个P. feriarum的样品是来自RRL法所提取的(ECM7144and ECM7210; 补充表5)。
在不同物种间测试候选基因
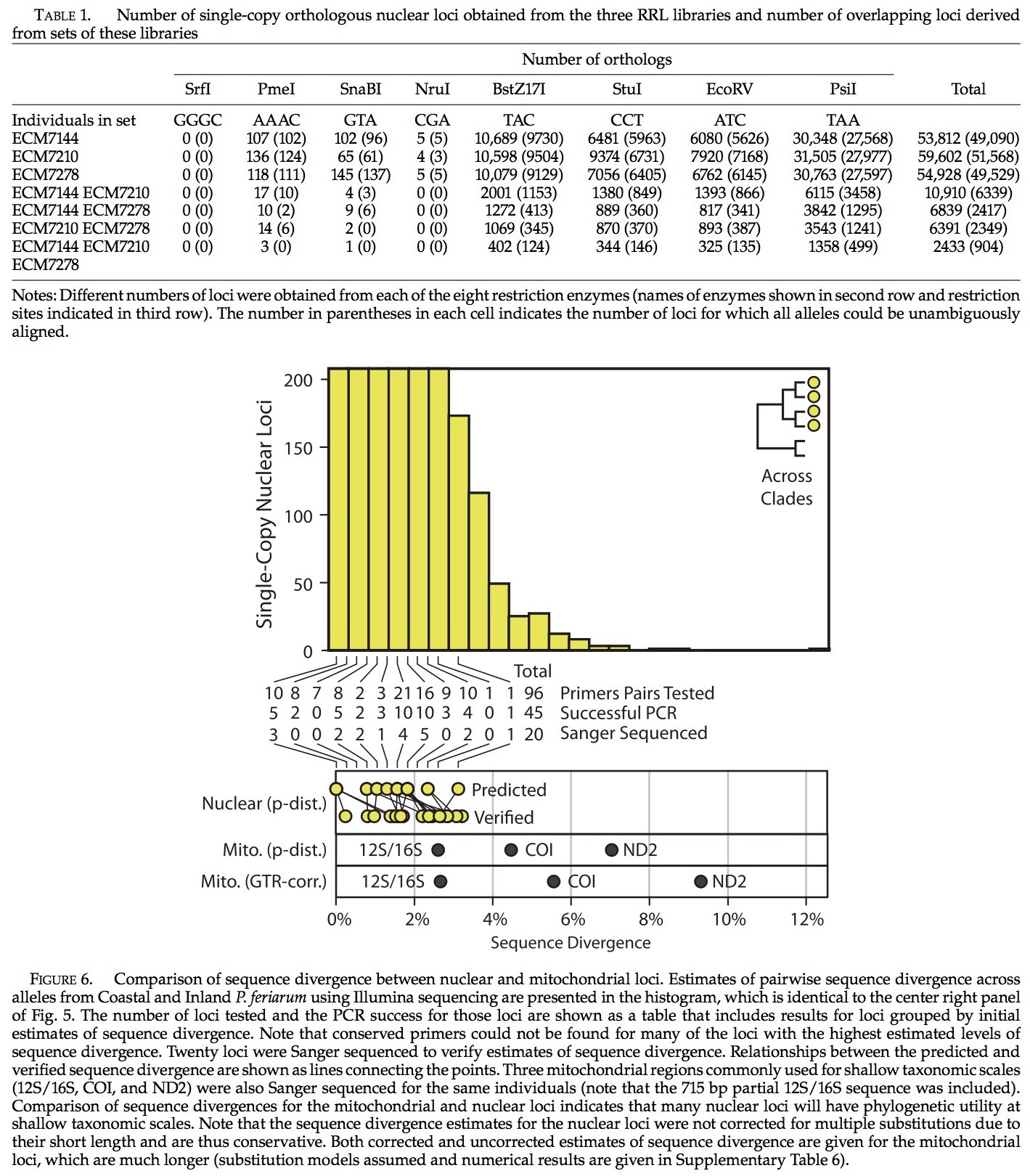
为了测试候选基因在不同物种间的效用,我们用PCR扩增了187个基因位点。为了得到这组基因位点,PCR测试包含了多轮次的方法。首先, PCR仅在这些基因位点起源的两个物种中进行(ECM2731 P.maculata and ECM7145 P.feriarum;补充表5)。第二,第一轮成功扩增的基因在拟蝗蛙属五个附加种内进行检测(ECM2695 P.regilla [P. sierra sensuRecuero et al. 2006a,b], MHP8258 P.streckeri, ECM5914 P. crucifer,ECM7199 P. brachyphona, andECM5055 P. nigrita)。第三,通过第二轮检测的基因,在拟蝗蛙属的剩余十个种全部进行检测(ECM0151 P. cadaverina, ECM0140 P. regilla [P.hypochondriaca sensu Recuero et al. 2006a,b], ECM4375 P. illinoensis, ECM5956 P. ornata, ECM7001 P. ocularis,ECM0080 P. brimleyi, ECM1144 P. clarkii, ECM2293 P. fouquettei, ECM1064 P. kalmi, and ECM7221 P. triseriata)。最终,通过第三轮检测的基因,来自两个hylid外群的两个个体被检测(LNB326 and ECM3184 Hyla cinerea, ECM5835 and ECM5938 Acris gryllus; Supplementary Table 5)。2,3,4轮的检测包括第一轮的检测的严格控制(ECM7145 P. feriarum)。
PCR反应包括1X Go TaqReaction Buffer (Promega),0.08 mM dNTPs, 0.4 U Go TaqDNA 聚合酶,每种引物0.2 M,以及总量10L的12ng模板DNA用于筛查或总量25L的30ng的模板DNA用于PCR扩增序列等过程。在康达德DNA发动机四分体2热循环仪上进行扩增时,使用下列过程:在95◦C条件下进行1循环 2分钟,在95◦C的条件下进行35循环30秒,引物在特定退火温度下处理30秒,(45, 47, 48, 49 or 51◦C),然后在72◦C条件下处理1分钟,然后在4◦C下停止(held)。PCR产物在120V的1%琼脂糖1× TAE下电泳20分钟并使用紫外线产品(UVP)荧光分析仪将结果可视化。在描述种内的测试时,如果扩大单一条带在我们所期待的大小范围,表明所给个体的基因位点成功分开,。我们将测序成功扩增的位点作为未来研究的一部分。在Dryad数据库测试所有位点的引物序列(http://datadryad.org, doi:10.5061/dryad.vh151q1c)。
估计序列分歧的水平
最后的评估基因的序列分歧水平有三类:个体内,个体间,种间。以上所采用的Illumina测序方法产生基因序列每个末端为70bp(位置序列在430bp之间)。我们采用序列分异的程度去观察每个序列基因的末端(每个基因140bp),作为评估整个序列基因分异的方法(570bp)。这个预测的准确性可能会受到两个因素的影响。首先,由于在全部个体中获得序列的限制位点和限制位点间的距离会在个体间得以保留,所以在个体间或者种间使用RRL法获取的基因,在所有基因组中可能会有偏差。从这一因素可能导致对序列分歧估计的负面偏差,我们希望我们的结果中属于高度变异的序列位点的数量是较少的。第二个因素是异质率。在这一因素下产生的偏差是在该规模下变异率的函数,当其在基因长度的规模下变化会产生最大程度的偏差是可以预期的。当在570bp基因位点末端的70bp与基因位点430bp的中心时,这种变异率有本质的差别。在这种情况下,基于末端的评估是在基因位点间的变化可以认为是正偏差。
为了要测试这种偏见并设立一个修正系数,我们对2个拟蝗蛙属个体一套基因位点(20)的整个长度进行了测序(Coastal clade: CM7144, Inland clade: ECM7210)。通过对这些相同个体进行Illumina测序后,我们可以在基于140bp(Illumina测序出末端序列)以及全部的570bp,(sequenced by Sanger)对序列的变异评估进行直接的比较。我们绘制了Illumina的评估与相应的Sanger评估进行了对比,检测一种明显的相关性,并且使用最适当的线性回归斜率以做出正确的估计。桑格测序包括以下过程:(1)从序列变化范围间选择20个基因;(2)使用QIA快速PCR纯化工具纯化这些基因的PCR产物,并且用50L分子级H20进行洗脱;(3)使用NanoDrop ND1000分光光度计将产物的浓度调整为10 ng/L。(4)测序使用Big-Dye v. 3.1 terminatorchemistry,佛罗里达州立大学的DNA测序设备Applied Biosystems 3730 Genetic Analyzer 进行测序。所有的桑格测序结果都被存放在Dryad(http://datadryad.org,doi:10.5061/dryad.vh151q1c)。
结论
序列分异
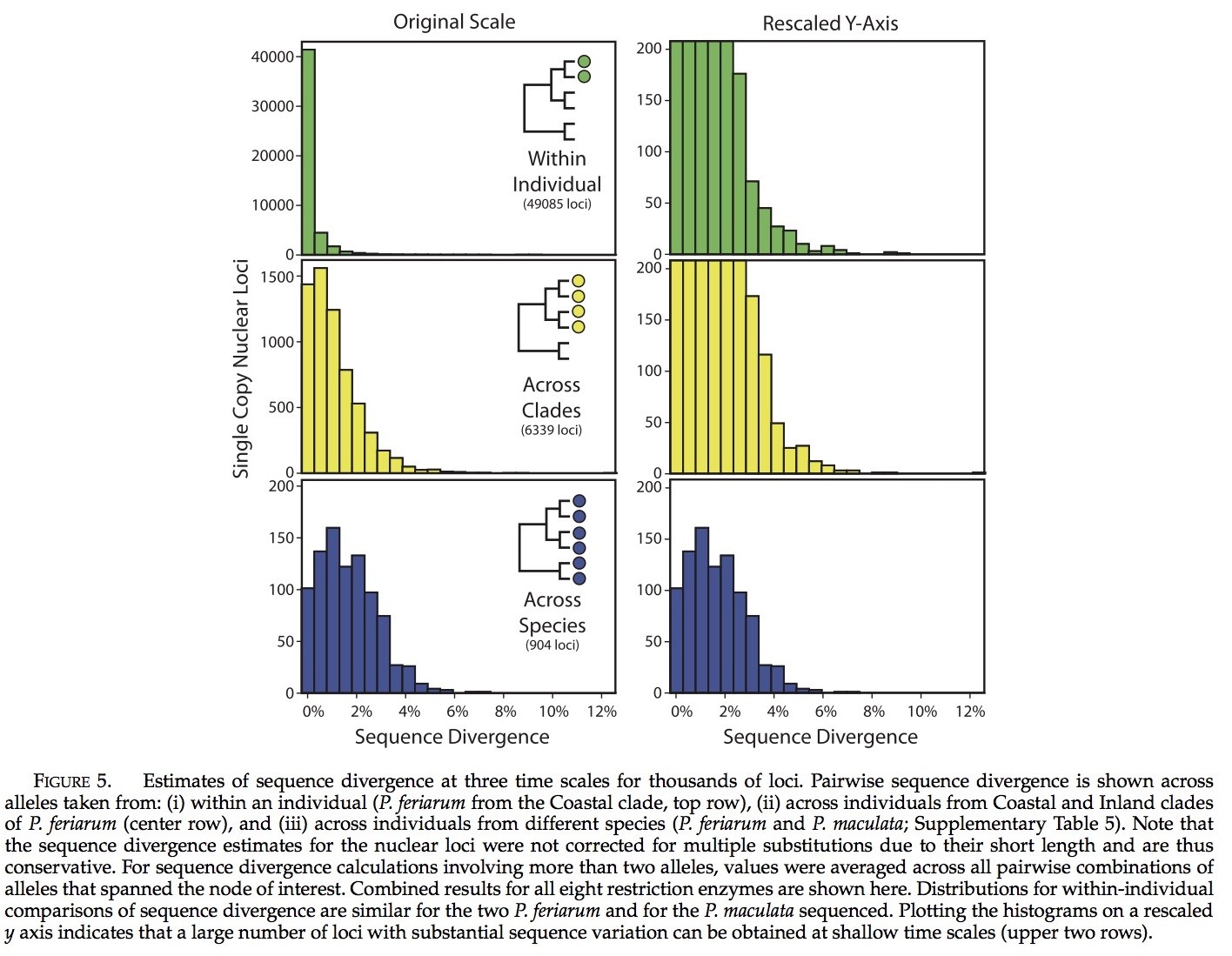
我们使用RRL测序法识别了超过100000个单次拷贝的核基因位点。大量的基因位点在浅层类水平上表现出高度的序列分异。序列分异在基因位点间的分布(图5)阐明了:虽然序列分异只是些许存在的(个体内有0.1%,拟蝗蛙属分支间有1.1%,种间有1.8%),但是通过评估发现:在这三个水平上大量基因位点存在高于3%的序列分异。在个体间,分支间,以及种间,我们分别了获取了195,419和146个基因位点对比的数据,发现其序列分异评估高于3% (ECM7144),。我们发现基因位点在种内的分异要高于种间分异(419 vs. 146),这可能是由于在种内许多基因位点与两个物种间的3个体中的基因位点相比,我们可以获取7倍以上的基因位点(6339 vs. 904; 表1)。回顾从两个物种中获取的大量基因位点,希望可以减少物种间进化的距离,因为限制位点侧部的基因与直接相关的个体可能存在差异(见下文),同时也是分割的标志。源自RRL方法所获取的序列,与H. japonica线粒体基因组序列相比,发现没有我们所获取基因位点在线粒体中并未发现。
RRL法可以用来获取对系谱地理学有益的序列分异水平上核基因位点。为了从细胞核基因位点来观察序列分异水平,我们计算现有序列的分异以便于在浅层次系统发育的水平上对三个线粒体区域进行共同的利用:12S/16S, COI和ND2。对于这些基因位点和通过桑格测序得到的全部核基因位点的序列分异在图6和补充表6中可见。大量核基因位点在序列分异其在比线粒体区域能更好的进行鉴别(图6)。我们获得了来自P. feriarum不同分支的两个个体的6339个未进行识别的基因位点,并且分别评估了比12S/16S, COI, and ND2更高序列分异的729,91以及6个基因位点。然而,对于许多基因评估有着更高的序列分异水平的引物既不能为基因位点所设计,也不能在P. feriarum个体中扩增(请注意,简并引物未被考虑在内)。通过桑格测序所得到的20个基因位点中,我们获得了6个比12S/16S分异度更高的序列。未经过桑格测序的基因位点比起COI 或者 ND2有着更好的分异度(在线粒体中基因位点序列分异的数量变化仅仅是由于位点间变异率,因为它们所继承的单一非重组块的影响)。从两个分支中所获取的基因位点的数量,通过评估发现仅仅有0.8%的是拟蝗蛙基因组的单次拷贝所得,估算发现发生在核细胞基因中的89,112, 11,124,和733的单次拷贝的基因位点,分别比12S/16S COI, 和ND2有着更高的序列分异度。这些结果使用双端测序法进行标记(不校正为多次替换)。
通过对末端片段的测序所得到序列分歧的估计,可用于促进核基因位点的理想序列变异的水平的探究。基于从双端测序的140bp和基于桑格~570 bp全长基因的序列(r =0.655, P = 0.001709),我们观察到序列变异的估计存在显著的相关性。桑格测序基于Illumina的评估的线性回归方程,当y轴截距时被设置为零时,产生的斜率为0.72。事实上,正如预期的那样,线性回归的斜率是1,表明基于140bp序列变异的估计在某种程度上存在正相关性(see Methods section)。通过将Illumina测序所有初步结果的估计值乘以0.72,我们已经纠正了偏差(见方法部分),从而确保纠正后斜率估计值为1.0。对所有的20基因位点序列分歧的估计(桑格测序,未校正的Illumina,已校正的Illumina)已在补充表6中给出。
通过测序错误所导致的序列分歧估计是无偏差的。重新考虑在基因位点的位置与相应片段的位置(补充图1)。因为平均测序错误的增加是从5’ 到3’进行读取的(补充图1),如果偏差是由于测序错误导致的,那么多态性的估计,会高度趋向于基因位点的中心。当包含了SNP基因位点的比例绘制成位点位置的函数时,我们并未发现有明显的趋势(图7)。相反,多态性的程度在位点间是均一化的,除了在读取开始端和结束端3bp的相关序列外,该序列几乎没有表明多态性。这个图示是我们所期待的,因为我们可以移除这些位置上含有多态性的基因位点以确保对齐的准确性(见方法部分)。当我们使用全部100bp的片段,试图整合读取和估计分异度时,我们观察到了一些偏差。由于此原因,我们的全部结果都基于对70bp序列对的分析。
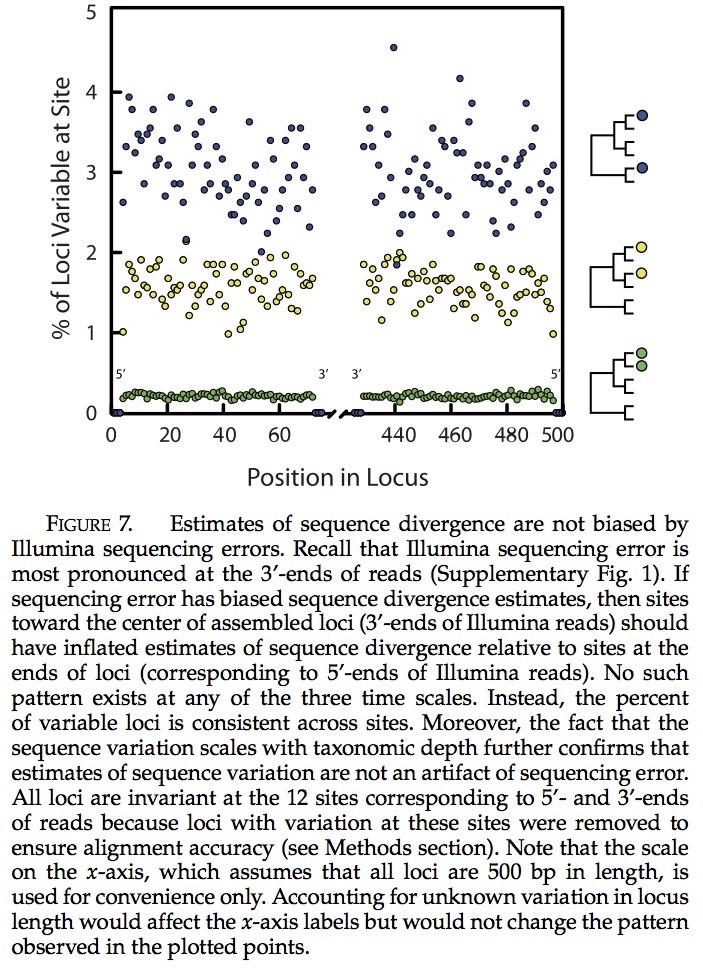
RRL法在物种间的效率
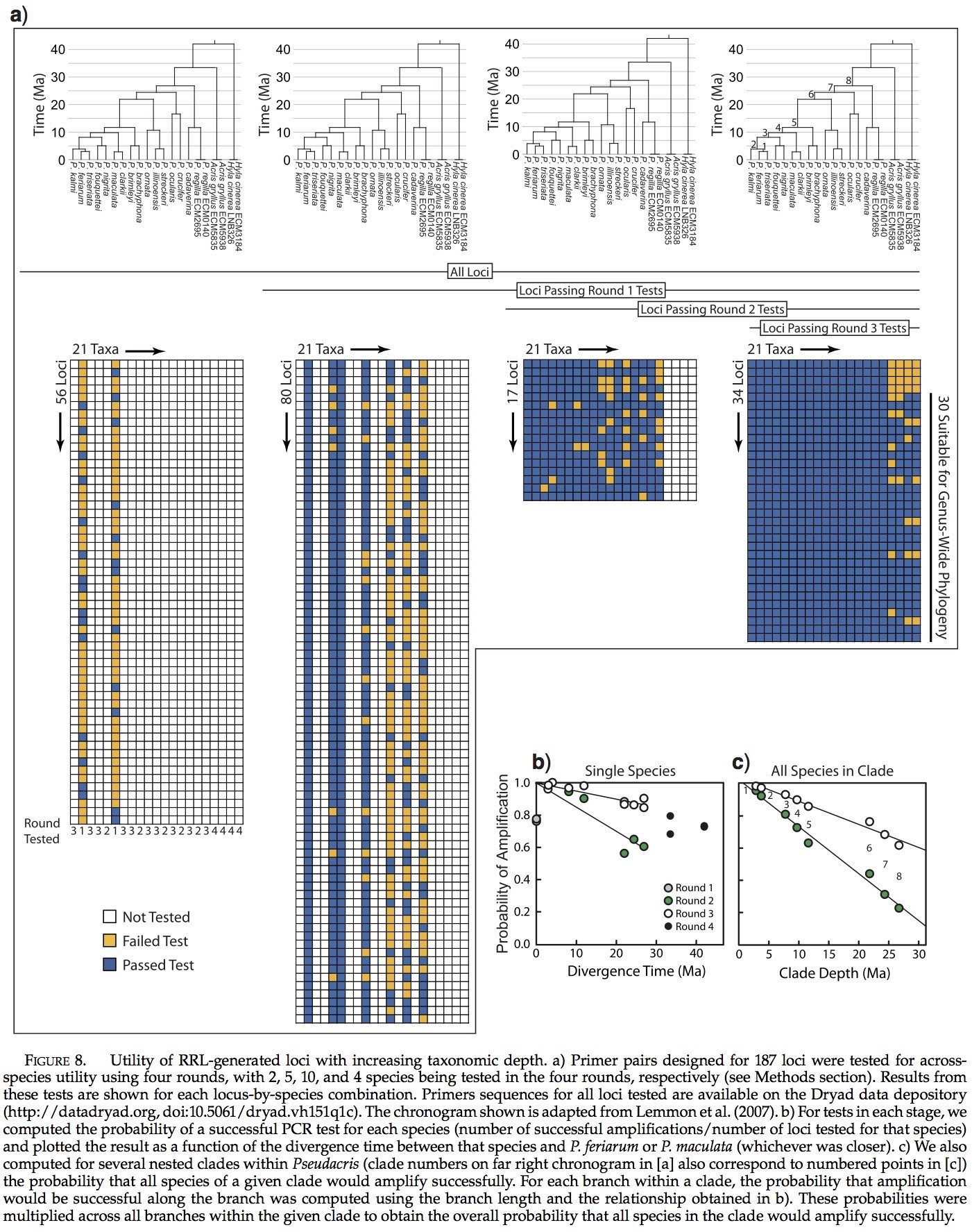
RRL测序法可以在物种内和物种间产生大量的基因位点。随着分类深度的增加,可用位点的数量稳步减少,我们给出了在多个物种的187基因引物筛选的结果(图8)。然而,我们可以获取30个基因位点(16%),以用PCR法成功将其在拟蝗蛙属全部17个种以及至少一个外群种(Acris gryllus or Hyla cinerea )中成功扩增。这些分支所包括的物种起源在30 Ma之前。如果仅是拟蝗蛙属的成员,那么大量的基因位点是可用的,34 (18%)(拟蝗蛙属的起源被认为是27 Ma; Lemmon et al. 2007)。我们对拟蝗蛙属内96个基因位点进行检测,发现其中51个得到了成功扩增(53%)。其他分支的相关可用基因位点数在图8c中给出。
PCR法显示的质量需要获取最终30个基因位点,这是很重要的。使用4轮显示法(见方法部分),我们进行了1675次PCR测试,其中成功了1339次(80%)。多轮检测的方法使我们避免了3927次可能检测中57%的检测(187基因位点× 21个体)。该方法的全部效率为32%,这可以通过除以PCR产物的总数来计算,在所有PCR测试后(1675,见以上)所得到的这些产物可以建立一个完整的数据矩阵和一个外群(30个基因位点×18人=540)。如果我们测试了所有可能测试3927,那么效率只有14%。这表明我们使用的多轮次的方法在效率上是将其中所有物种的全部基因测试的全部测试法的两倍以上。另一种方法是测试最远的两个相关的物种中第一个物种的扩增(例如Pseudacris feriarum and Hyla cinerea ),然后在剩余物种中扩大加以验证。基于我们收集的数据,很难确定这种方法的相对效率。
实验设计的效率
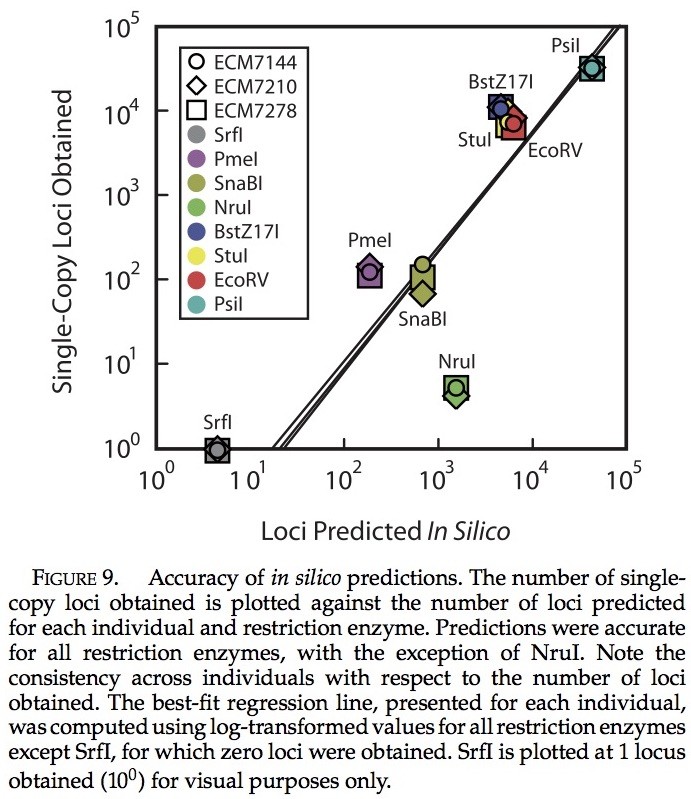
使用热带爪蟾基因组进行生物信息学预测,给大多数限制酶的基因位点的数量提供了合理的评估。我们在生物信息学基因位点预测与实际获得的单次拷贝数量间发现了很强的相关性(图9)。限制性内切酶预测产量的一小部分产生少量的单次拷贝基因位点(e.g., SrfI)。反过来也是如此(e.g., PsiI)。唯一的例外是NruI,产生了的基因位点数目大幅度减少(可能由于其甲基化敏感性)。每个个体所获得的单次拷贝基因位点总数与预测数惊人的接近(与61,261相比,预测数为53,812 来自P. feriarumECM7144, 59,602 来自P. feriarumECM7210, 以及 54,928 来自P. maculataECM7278)。
多种因素导致序列读取的某种低效利用。第一个因素是读取的质量。我们严格的质量要求(每次读取基于第一个70bp其最质量分数最小值为20)使我们去除了45%的片段。5'端匹配读取的酶切位点还导致大量读取部分的去除(39%)。合在一起,这两个因素导致我们在序列读取时损失了65%的片段。其他降低效率的主要因素是多拷贝基因的存在(例如,重复的位点)和重复的元素(例如,短元素穿插[SINEs]长散置元[LINEs])。最初的基因位点(集群>10倍范围,表现为单拷贝基因,多次拷贝基因和重复元素)包括了98%的通过了质量过滤器的片段,然而在最后的单拷贝基因中,只有31%的片段通过了质量过滤器。计算所有步骤,只有11%的原始读取可以用来建立最终的等位基因位点。虽然这些因素似乎没有使基因位点数目大大减少(相对于生物信息学预测),观察可知他们确实有助于大幅减少每个位点的平均覆盖范围:覆盖范围为每个位点979片段,但是观察到每个位点只有65个片段(93%的片段消失了;补充表2-4)。我们计算随着每百万对质量筛选的片段数中单次拷贝基因位点的数量,发现限制性内切酶的效率在各种限制酶中变化十分大,从0 (SrfI)到2693(PsiI)。这种级别的变化表明,使用一些限制性内切酶可能比其他更能节约成本。然而,这种效率措施将在基因组中自发变化并且其大小范围选择在库的准备期也会发生改变。
讨论
高通量核基因位点在系谱地理学和系统发育中的发现
本文提出了一种新的方法来识别成千上万的核位点,以便我们研究在浅层次系统发育和系谱地理学上的问题。这种方法的发现使得研究人员能够克服在这些领域的两大挑战(Avise 2009): 增加含有信息的单次拷贝核基因位点的数量和有效的等位基因。我们对序列分歧分布的估计表明,这些基因位点中许多将在浅层分类尺度下提供有效信息(~7% 的基因位点在种内分异 >3% )。此外,在更深的分类尺度下基因位点的PCR检测表明:跨物种间基因位点扩增的数量随着分类深度的增加逐渐减少,只有>16%的目标基因可以在所有物种内得以扩增。因此,我们期望对这些基因位点研究是有有所收获的,包括从属内到种内水平的系统发育分析。我们可以降低一些严格的质量控制措施的标准,来获得额外基因位点,如仅使用不包含空白的对齐的基因位点。此外,使用更长的片段(e.g., paired-end 250 bpreads expected on the Illumina platform in the third quarter of 2012)可以使多个基因引物设计具有很高的序列变异的水平。理想情况下,额外的基因位点识别的验证需要阐明等位基因的孟德尔遗传,基因位点之间的自由组合,在基因位点内的低重组率,以及每个基因位点选择性的中立性。
通过本方法识别在物种内和物种间的基因位点得以成功,是由于我们所使用的实验设计。多倍体的利用允许我们:(1)确定跨物种的基因位点的发展以识别保留的引物区域(2)估计序列变异的水平,以便在种群内位点识别的基因位点选择的灵活性。此外,在高覆盖率下使用双端测序使得我们可以识别单次拷贝的基因位点以及基因位点内的等位基因。虽然最后这些基因位点的测序还利用双端,高覆盖率的方法,可以使用PCR进行扩增,并且碎片可以进行限制吸收和大小选择。另外,一个可以应用基于RRL的丰富度方法讨论了这里的所有样品和放弃基因位点的筛选以及PCR检测程序。应用基于RRL的丰富度方法去检测所有样品的缺点是它可以很大程度的减少基因位点的数量,这些位点可以在相同覆盖范围内进行测序,就像将在扩增序列中测得的那样。最佳方法可能取决于研究中样本的数量,也就是我们是否需要一大批多态性较高的基因位点。
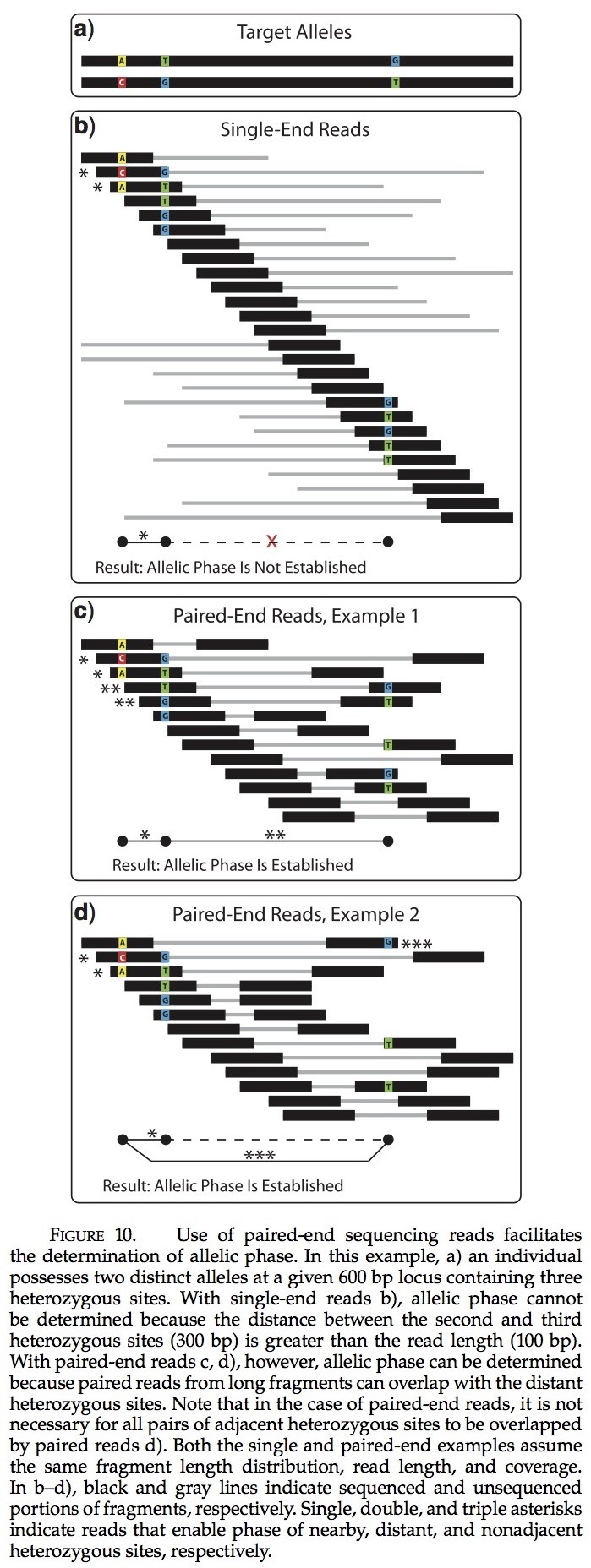
在一些环境中,读取比基因位点长度更短的片段阻碍研究员建立基因相的能力。在此,通过单个杂合个体,我们关注起决定性作用的两个等位基因序列,利用该个体进行序列读取(作为反对来自第二阶段的序列数据; Stephens et al 2001)。为了建立等位基因对,必须满足两个条件:(1)必须确定杂合子位点(2)杂合子位点的特征状态之间的关系必须建立。使用这项研究中使用的建模方案(以及假定的序列错误率),鉴别杂合位点必须采用10倍的最小覆盖范围。满足第二个条件需要通过读取两个或更多重叠的位点,使所有杂合子位点之间形成连接(直接或间接的)。在单端测序的情况,必须至少有一个片段与每对相邻杂合子位点重叠(更可能需要以适应测序错误)。如果任何两个相邻杂合子位点之间的距离大于片段的长度,然后无法确定等位基因的状态。在杂合子位点之间的距离较小的时候,建立具有高度的序列变异位点的等位基因更容易。当序列变异水平较低时,更长的片段是必需的,这样可容纳较大的杂合子位点间距。然而,使用来自双端测序片段中的信息可以规避对长片段的需求(图10)。双端测序的片段,如果他们重叠连续片段的任何一个或两个不同的片段来自同一对,那么在一对杂合子位点的等位基因的特点可以得到。如果任何两个相邻杂合子位点之间的距离大于库中准备的片段的长度,然后无法确定等位基因的状态。在双端测序的情况下,片段长度和碎片长度决定建立等位基因的可能性。通常,来自片段中长度分布较广的库的准备可以最好的进行。在补充表2中,我们目前模拟结果表明,双端测序片段优于单端片段,其他也一样。
未来位点辨识RRL方法的建议
从本研究中获得的经验使我们能够给研究人员做出一些推荐,以优化他们使用RRL法去挖掘基因位点的效率。这种方法的一个明显的局限性是:对于每个专门的研究系统,必须专门挖掘基因位点。然而,本节的目标是使用我们所学到的方法将测序的成本最小化并将基因位点识别步骤效率最大化。
限制性消化和生物信息学的预测。——我们的经验表明,不同差异的参考基因组可以用来估计基因位点的数目,从而预测从一个特定的限制性内切酶和插入大小的组合(图3;补充表2-4)。核苷酸组成的标记可对可获取基因位点的数目做一个预测(见补充表3)。我们的估计是合理精确的,这是基于在热带爪蟾的生物信息学预测(源于20亿年前;来自拟蝗蛙属的分异),(图9;补充表2-4)。这个结论下,在未来的研究使用这种方法可以使用更少的限制酶。我们所检测出最高效的限制酶是EcoRV, PsiI, 和SnaBI, 在每百万质量筛选的片段中,每个可以产生大约2000个单次拷贝的核基因位点,可以得出我们所利用碎片的大小范围。一种限制酶NruI,产生的位点比预期要少,每百万质量筛选片段只能产生10个基因位点(补充表2-4)。因为NruI可能对甲基化敏感,所以NruI的低效率可能是由于不完全消化。我们发现它是在基因位点的识别效率较低,所以我们建议避免使用这种酶。因为基因组的特性可能会影响识别效率,所以目的是识别较长基因位点的研究人员应相信我们使用最密切相关的物种基因组资源研究所得到的在生物信息上的发现。
一定要注意的是,除非采取某些预防措施,否则使用一种单一的酶可能产生劣质数据序列。原因是因为:目前的Illumina软件校准基于我们读取时所使用的几个基底(Davey et al. 2011)。对于这些基底浅层序列分异度的校准(作为将采用一个单一的限制酶)可能因此产生劣质的序列数据。因为汇集了来自八种限制性内切酶库,我们成功地避免了这个问题。通过源于整个基因组DNA的一个库与RRL方法相混合,研究人员倾向于使用少量的限制酶来规避这个问题。我们已经成功的从一种单一的限制性内切酶生成序列的RRL库,通过混合它与整个基因组库的比率为75:25(RRL:genomic DNA; Lemmon and Lemmon,unpublished data)。
我们在生物信息中对每个位点的覆盖率预测过于乐观。,通过预期片段对的总数除以预期基因位点的总数,我们计算覆盖范围的期望值。为了我们的实验设计,其中包括由八种限制性酶消化所得的碎片集合,并且选择600–720 bp碎片,每个基因位点我们预测979倍的覆盖范围。单次拷贝的基因位点实际平均覆盖率的观测值为67,预期覆盖范围只有7%(补充表2-4)。三个因素导致了这一覆盖范围的缺失。最大的因素是多次拷贝基因位点和重复元素的存在。这两类位点的去除导致68%的片段的缺失。由于这些元素导致的高缺失率可能并不令人十分惊讶,因为我们知道热带爪蟾的基因组大小只有拟蝗蛙基因组的大小的39%,该模式生物常用于预测覆盖范围。通过列入修正模型和目标物种之间的基因组大小的差异的因素,未来在生物信息中对覆盖率的预测可以改善。导致覆盖下降的第二大因素是劣质片段的存在。因为我们需要准确估计序列分异,我们使用一个非常严格的质量筛选器,导致去除了45%的片段(补充表2-4)。虽然通过使用不太严格的质量筛选器,研究人员不感兴趣的序列变异水平可以提高应用的覆盖范围,这个因素很难得到解释,因为片段的质量可能有在库的准备和测序过程中发生改变。第三个因素导致覆盖率大幅下降的因素是有5’序列与八种限制性内切酶中的一个不匹配。这可能是在库准备之前,不匹配碎片意外破碎,导致39%的片段发生缺失。未来的库的准备改进或使用更高质量的DNA可以减少损失。总之,这三个因素导致相对于预期的覆盖率89%的损失。最后一个因素也可能对覆盖范围的估计并不准确,这是由于大小选择产生的在片段的长度内覆盖范围的峰值分布,而我们的生物信息分析认为覆盖率的分布是均匀。这一因素导致在基因位点间的变异比预期的高。尽管原始片段的利用率较低,但基于我们所使用高通量测序平台的特性,我们可以挖掘大量的单次拷贝的基因位点,。
库的准备和测序——库的准备至少在两个方面可以进行改进:第一,增加选择片段的大小范围的宽度,可以增加获得个体或物种同源基因的数量。其原因在于,更宽的范围会增加容纳位点长度的范围(由于存在插入或缺失的)。当然,更多的片段必须获得同样的覆盖水平,除非个体或基因位点的数量减少了。增加大小范围的宽度也可能对基因位点间覆盖率的变异的减少产生有益的影响。第二种可能提高库的准备的方法是选择较大的破碎片段,这可以产生更长的基因位点。在我们实验室,我们在没有对库的准备和测序协议进行修改的状况下,通过Illumina平台进行100bp成对双端测序,已成功测序片段的末端长度增加到900bp。(Lemmon et al. 2012)。其他研究人员将序列片段长度提高至2kb(e.g., Li et al.2009)。
通过一些测序上小小的改变以及我们所采用引物的筛选策略,可以降低产生核基因位点的成本。最明显的修改是只使用一条测序路径。自从这项研究的数据收集后,Illumina测序公司发布了新试剂(v3)能够在每条路径生成3倍以上的数据。因此,对于那些打算使用一个相似的实验设计和基因组的大小的研究人员来说,一个Illumina碱基读取路径应该可以产生类似的覆盖范围。然而,需要注意的是,库需要通过个体集中后才能够索引化或条形码化。索引,其中一个特定的寡核苷酸设计示例在一个单独的序列片段中是适配和测序,将成为使用条形码将每个片段可用寡聚核苷酸减少后的首选(寡聚核苷酸样品作为一个正常序列片段5’N端的基底)。第二个修改降低成本是只采用了对~70 bp末端进行双端测序。尽管我们对100 bp末端进行双端测序,由于担心这种低质量的区域影响我们的序列变异的估计,我们放弃了每个片段从3′末端的30bp。第三次修改是对更少的个体进行测序。如果基因位点的识别只需要在种群内使用,那么可能不需要包括来自多个物种的个体。此外,如果基因位点是随机而不是需要增强的序列变异的水平,那么对多个个体排序可能是多余的。减少测序个体的数量将使大量待开发的位点在相同水平下得以发掘,但会妨碍PCR效率,因为保守区域可能不容易进行识别。一个测序平台产生较小的片段数,如Illumina MiSeq也可以利用以降低每次的运行成本。目前这个平台的输出产生大约1200万个片段。例如,基于我们的限制酶EcoRV效率的估计,这意味着使用一个个体我们可以获得大约2万个基因位点和一个比我们这里使用的更大的尺寸范围。降低PCR试剂和劳动力成本,发展多重PCR检测,以允许多个基因位点在相同的反应下得以扩增(e.g.,Meuzelaaret al. 2007)。软件或服务的多重设计可从诸如PREMIER Biosoft公司获取(PrimerPlex软件)。
解释序列分异的评估
我们研究的目标之一是估计整个核基因位点序列变异的分布。但是,在解释时,对这些估计必须加以关注。原因是对于每个所给基因位点序列的分异度是序列进化分异率的一个函数,这种进化是基因位点和等位基因联合的随机性质所决定的(Hudson 1991)。自从我们在浅层次时间尺度上进行工作和在每次比较中使用了少量等位基因后,我们预期观察到的变异某些部分是由于联合的随机性。选择具有高度的序列变异的基因位点可能使基因位点偏向于在更深的时间下进行联合,因为从更深的合并将有助于增加序列变异。如果模型对下游进行分析需要无偏差的基因位点,那么我们可以采取两种方法以避免这种偏差。第一种方法是显而易见的:在序列分异情况下随机选择基因位点。这样,基因位点是一个更随机的基因组的代表,虽然由于需要保守的引物区域,其结果并不可能完全随机。第二种方式来减少这些影响包括在更多的个体中进行基因位点的识别。增加个体的数量将允许序列分歧评估下的变化,它可以更准确地反映相对的进化率,因为更多的相联系的事件可以表现并且可以缓解随机效应。
总结
这项研究阐述了一种快速、低成本RRL方法,来产生大量的高变异度的核基因位点,以便于我们对任何二倍体系统进行系统发育和系统地理学方面的研究。这种方法对在具有很少或没有基因资源的非模型系统条件下工作的研究人员尤其有用。在最近参照基因组中的生物信息预测优于经验性工作,这为从这种方法中产生预期基因位点数目做出了合理地评估,并且促进了需要获取充足覆盖的测序水平的估计。在从多个分类单元检测目标物种来设计基因位点引物时,正如预测的那样,PCR扩增的成功的同时,进化变异水平降低。尽管这一趋势存在,大量可变的基因位点可以被识别,并在整个目标属得到扩增。因此,对于需要适度的大量核基因位点的系统发育学和系谱地理学的研究,这一方法是有用的。
SUPPLEMENTARYMATERIAL
Supplementarymaterial, including data files and/or online-only appendices, can be found atwww.datadryad.org at doi:10.5061/dryad.vh151q1c.
FUNDING
This work was supported by Florida StateUniversity new faculty set-up funds to A.L. and E.M.L.
ACKNOWLEDGEMENTS
We are especiallygrateful to Lisa Barrow, Hannah Ralicki, and Sandra Emme for conducting the PCRscreening and Sanger sequencing for this project. We thank Brian Caudle,Mallory Bedwell, and Sandra Emme for general laboratory support. We are gratefulto David Cannatella and Travis LaDuc of the Texas Memorial Museum at theUniversity of Texas, Austin, and to Curtis Schmidt and Joseph T. Collins of theSternberg Museum of Natural History, Fort Hays State University, for tissueloans. We also thank Brooke Aden, Lisa Barrow, Joseph T. Collins, David Hall,Chris Hobson, Andrew Landis, . Bruce Means, Moses Michelsohn, Cameron Siler,Courtney Swisher, and Mitch Tucker for assisting with tissue collection.
REFERENCES
Akaike H. 1974. A new look at statistical model identification. IEEE Trans.Automat. Control 19:716–723.
Altshuler D., Pollara V.J., Cowles C.R., VanEtten W.J., Baldwin J., Linton L., Lander E.S. 2000. An SNP map of the humangenome generated by reduced representation shotgun sequencing. Nature407:513–516. Avise J.C. 2009. Phylogeography: retrospect and prospect. J.Biogeogr. 36:3–15.
Baird N.A., Etter P.D., Atwood T.S., Currey M.C., Shiver A.L., Lewis Z.A.,Selker E.U., Cresko W.A., Johnson E.A. 2008. Rapid SNP discovery and geneticmapping using sequenced RAD markers. PLoS One 3:e3376.
Barbazuk W.B., Bedell J., Rabinowicz P.D. 2005. Reduced representationsequencing: a success in maize and a promise for other plant genomes. BioEssays27:839–848.
Brito P.H., Edwards S.V. 2009. Multilocus phylogeography and phylogeneticsusing sequence-based markers. Genetica 135:439–455.
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., MaddenT.L. 2009. BLAST+: architecture and applications. BMC Bioinform. 10:421.
Davey J.W., Hohenlohe P.A., Etter P.D., BooneJ.Q., Catchen J.M., Blaxter M.L. 2011. Genome-wide genetic marker discovery andgenotyping using next-generation sequencing. Nat. Rev. Genet. 12:499–510.
Edgar R.C. 2004. MUSCLE: multiple sequence alignment with high accuracy and highthroughput. Nucleic Acids Res. 32:1792–1797.
Emerson K.J., Merz C.R., Catchen J.M., Hohenlohe P.A., Cresko W.A., BradshawW.E., Holzapfel C.M. 2010. Resolving postglacial phylogeography usinghigh-throughput sequencing. Proc. Natl. Acad. Sci. USA 107:16196–16200.
Filarczyk A., Nadachowska K., Hofman S., Litvinchuk S.N., Babik W., Stuglik M.,Gollman G., Choleva L., Cogalniceanu D., Vukov ˇ T., Džukié G., Szymura J.M.2011. Nuclear and mitochondrial phylogeography of the European fire-belliedtoads Bombina bombina and Bombina variegata supports theirindependent histories. Mol. Ecol. 20:3381–3398.
Galtier N., Nabholz B., Glemin S., Hurst G.D.D. 2009. Mitochondrial DNA as amarker of molecular diversity: a reappraisal. Mol. Ecol. 18:4541–4550.
Glenn T.C. 2011. Field guide to next-generation DNA sequencers. Mol. Ecol.11:759–769.
Hare M.P. 2001. Prospects for nuclear gene phylogeography. Trends Ecol. Evol.16:700–706.
Hudson R.R. 1991. Gene genealogies and the coalescent process. Oxf. Surv. Evol.Biol. 7:1–44.
Hurst G.D.D., Jiggins F.M. 2005. Problems with mitochondrial DNA as a marker inpopulation, phylogeographic, and phylogenetic studies: the effects of inheritedsymbionts. Proc. R. Lond. B Biol. Sci. 272:1525–1534.
Igawa T., Kurabayashi A., Usuki C., Fujii T., Sumida M. 2008. Completemitochondrial genomes of three neobatrachian anurans: a case study ofdivergence time estimation using different data and calibration settings. Gene407:116–129.
Jennings W.B., Edward S.V. 2005. Speciational history of Australian grassfinches (Poephila) inferred from thirty gene tree. Evolution59:2033–2047.
Kerstens H.H.D., Crooijmans R.P.M.A., Veenendaal A., Dibbits B.W., Chin-A-WoengT.F.C., den Dunnen J.T., Groenen M.A.M. 2009. Large scale single nucleotidepolymorphism discovery in unsequenced genomes using second generation highthroughput sequencing technology: applied to turkey. BMC Genomics 10:479.
Lemmon A.R., Emme S., Lemmon E.M. 2012. Anchored hybrid enrichment formassively high-throughput phylogenomics. Syst. Biol. 61:747–761.
Lemmon A.R., Lemmon E.M. 2008. A likelihood framework for estimatingphylogeographic history on a continuous landscape. Syst. Biol. 57:544–561.
Lemmon E.M., Lemmon A.R., Cannatella D.C. 2007. Geological and climatic forcesdriving speciation in the continentally distributed trilling chorus frogs (Pseudacris).Evolution 61:2086–2103.
Li R., Fan W., Tian G., Zhu H., He L., Cai J., Huang Q., Cai Q., Li B., Bai Y.,Zhang Z., Zhang Y., Wang W., Li J., Wei F., Li H., Jian M., Nielsen R., Li D.,Gu W., Yang Z., Xuan Z., Ryder O.A., Leung F.C., Zhou Y., Cao J., Sun X., FuY., Fang X., Guo X., Wang B., Hou R., Shen F., Mu B., Ni P., Lin R., Qian W.,Wang G., Yu C., Nie W., Wang J., Wu Z., Liang H., Min J., Wu Q., Cheng S., RuanJ., Wang M., Shi Z., Wen M., Liu B., Ren X., Zheng H., Dong D., Cook K., ShanG., Zhang H., Kosiol C., Xie X., Lu Z., Li Y., Steiner C.C., Lam T.T., Lin S.,Zhang
Q., Li G., Tian J., Gong T., Liu H., Zhang D., Fang L., Ye C., Zhang J., Hu W.,Xu A., Ren Y., Zhang G., Bruford M.W., Li Q., Ma L., Guo Y., An N., Hu Y.,Zheng Y., Shi Y., Li Z., Liu Q., Chen Y., Zhao J., Qu N., Zhao S., Tian F.,Wang X., Wang H., Xu L., Liu X., Vinar T., Wang Y., Lam T.W., Yiu S.M., et al.2009. The sequence and de novo assembly of the giant panda genome. Nature463:311–317.
Meuzelaar L.S., Lancaster O., Pasche J.P., Kopal G., Brookes A.J. 2007.MegaPlex PCR: a strategy for multiplex amplification. Nat. Methods. 4:835–837.Meiklejohn C.D., Montooth K.L., Rand D.M. 2007. Positive and negative selectionon the mitochondrial genome. Trends Genet. 23:259–263.
Neigel J.E., Avise J.C. 1986. Phylogenetic relationships of mitochondrial DNAunder various demographic models of speciation. In: Nevo E., Karlin S.,editors. Evolutionary processes and theory. New York: Academic Press. p.515–534.
Pyron R.A. 2011. Divergence time estimation using fossils as terminal taxa andthe origins of Lissamphibia. Syst. Biol. 60:466–481.
Rozen S., Skaletsky H.J. 2000. Primer3 on the WWW for general users and forbiologist programmers. In: Krawetz S., Misener S., editors. Bioinformaticsmethods and protocols: methods in molecular biology. Totowa (NJ): Humana Press.p. 365–386. Recuero E., Martínez-Solano í., Parra-Olea G., García-París M.2006a.
Phylogeography of Pseudacris regilla (Anura: Hylidae) in western NorthAmerica, with a proposal for a new taxonomic arrangement. Mol. Phylogenet.Evol. 39:293–304.
Recuero E., Martínez-Solano í., Parra-Olea G., García-París M. 2006b.Corrigendum to “Phylogeography of Pseudacris regilla (Anura: Hylidae) inwestern North America, with a proposal for a new taxonomic rearrangement”. Mol.Phylogenet. Evol. 41:511.
Smith S.A., Arif S., de Oca A.N., Wiens J.J. 2007. A phylogenetic hot spot forevolutionary novelty in Middle American treefrogs. Evolution 61:2075–2085.
Spinks P.Q., Thompson R.C., Shaffer H.B. 2010. Nuclear gene phylogeographyreveals the historical legacy of an ancient inland sea on lineages of thewestern pond turtle, Emys marmorata in California. Mol. Ecol.19:542–556.
Stephens M., Smith N.J., Donnely P. 2001. Anew statistical method for haplotype reconstruction from population data. Am.J. Hum. Genet. 68:978–989.
Thompson R.C., Shedlock A.M., Edwards S.V., Shaffer H.B. 2008. Developingmarkers for multilocus phylogenetics in non-model organisms: a test case withturtles. Mol. Phylogenet. Evol. 49:514–525.
Townsend T.M., Alegre R.E., Kelley S.T., Wiens J.J., Reeder T.W. 2008. Rapiddevelopment of multiple nuclear loci for phylogenetic analysis using genomicresources: an example from squamate reptiles. Mol. Phylogenet. Evol.47:129–142.
Van Tassell C.P., Smith T.P.L., Matukumalli L.K., Taylor J. F., Schnabel R.D.,Lawley C.T., Haudenschild C.D., Moore S.S., Warren W.C., Sonstegard T.S. 2008.SNP discovery and allele frequency estimation by deep sequencing of reducedrepresentation libraries. Nat. Methods 5:247–252.
Wiedmann R.T., Smith T.P.L., Nonneman D.J. 2008. SNP discovery in swine byreduced representation and high throughput pyrosequencing. BMC Genetics 9:81.
Zhang D-X., Hewitt G.M. 2003. Nuclear DNA analyses in genetic.studies ofpopulations: practice, problems and prospects. Mol. Ecol. 12:563–584.
Zink R.M., Barrowclough G.F. 2008. Mitochondrial DNA under siege in avianphylogeography. Mol. Ecol. 17:2107–2121.
https://blog.sciencenet.cn/blog-536560-1084811.html
上一篇:2017年学术年会“昆虫分类与区系”分组参会随想
下一篇:传粉者分布、生态功能以及保护措施的多样性