博文
衰老过程中端粒、线粒体及干细胞功能衰退的相关性
||
衰老过程中端粒、线粒体及干细胞功能衰退的相关性
Linking functional decline of telomeres, mitochondriaand stem cells during ageing
美国哈佛医学院德纳-法伯癌症研究所Ergün Sahin, Ronald A. DePinho
摘要
人类遗传病和突变小鼠模型的研究证据表明,基因组维护机制,DNA损伤信号和代谢调控三者间的协调,共同推动了老龄化的进程。特别是增龄性端粒损伤、端粒“加帽”(capping)功能削弱及相关的p53激活,被认为是组织干细胞功能衰退和线粒体功能紊乱的主要“元凶”(instigator),从而对各组织的更新和生物能利用造成不利影响。端粒、干细胞和线粒体,与调控基因组完整性的关键分子之间存在相互作用;而在相关模型的构建过程中,“干细胞属性”(stemness)和新陈代谢特征为揭示多因素促发衰老和增龄性疾病的机制,提供了模型框架。
Sahin E,DePinho RA.Linking functional decline of telomeres,mitochondria and stem cells during ageing.Nature. 2010,464,520–528
在过去一个世纪,医学的进步已将工业化国家的人口预期寿命延长了近一倍。长寿浪潮(longevity boom)的出现,主要是由于新生儿医疗的改善,传染病的预防和治疗以及心血管和内分泌疾病的干预;而预计到2025年,60岁及以上老年人口将达到12亿。医疗技术对老年人口健康和福祉的保障具有深刻的影响,人类愈加迫切地需要更全面揭示衰老及增龄性疾病的分子通路和生物学进程。
我们揭示衰老进程的细节总会面临如下的挑战,比如组织和有机体功能衰退的渐进性和异质性,衰老表型(phenotype)的复杂性和多样性,并且在分子和细胞水平,缺乏能够量化衰老程度生物标志物。更复杂的问题是,哺乳类的衰老进程受到外部因素(如饮食摄入)、内源性应激[如ROS、端粒损耗(erosion)]的影响;其他情况下,生殖细胞系变异可影响调控DNA修复的遗传因子[1,2]。从酵母到灵长类的多种生物模型研究提示,探索以下领域具有重要意义:①基于磷脂酰肌醇-3激酶[phosphatidylinositol-3-OH kinase,PI(3)K]通路的代谢通量(metabolicflux)分析;②主代谢调节因子(如sirtuins)的活性;③通过DNA有效修复及抗氧化机制保持基因组完整性;④端粒损耗或ROS过量所致的基因毒性应激(genotoxicstress)水平;⑤p53和p16抑癌通路的激活;⑥线粒体储备(reserve)和功能的稳健性(robustness);⑦正常情况下细胞外环境与细胞因子的整合性[2,3,4]。但目前尚不完全清楚,上述途径和进程如何共同促进或延缓了衰老的进程。
越来越多的证据提示,推动衰老、干细胞储备凋亡性损耗,以及增龄性组织变性的核心要素是端粒和p53介导的DNA损伤信号。这种细胞检测点(checkpoint)机制可促进高度增殖组织的功能衰退;在衰老过程中,该机制同样对静态(quiescent)组织(如心、脑、肝)产生破坏效应,但要对此做出合理解释则更加困难。本文中我们提出一个假设模型,可将端粒损伤和p53激活与干细胞和线粒体功能紊乱联系起来。虽然机体各组织类型不同、增殖特征各异,但上述模型可针对端粒如何影响衰老机体健康这一命题,提供一个统一的解释,
衰老表型
尽管衰老确切的分子机制尚未明了,但仍可给出衰老表现作基本的界定,即功能程度的整体衰退,表现在各器官不能保持基线的组织稳态,不能对应激性生理需求做出适当反应[3,5]。在多种衰老组织,上述衰退呈渐进性和适度性,但到晚年则加速;在机体为防御各种应激源(stressor)而作出强烈的生理反应或再生反应(regenerativeresponse)的情况下,这种衰退尤其明显。在解剖和生理水平,组织的细胞结构(cellularity)破坏、再生能力下降,似与许多典型的增龄性病症(如肌肉萎缩、贫血,免疫功能低下、创伤愈合障碍)密切相关(框1)
与功能衰退进程同步,年龄是慢性疾病进展的重要危险因素。在美国65岁以上人群中,近50%罹患心血管疾病,35%为关节疾病,15%为2型糖尿病,10%为肺疾病。其中某些病症可发生严重的功能衰退,并造成显著的社会和经济损失。脑卒中和痴呆是65岁以上人群长期住院最常见的原因,仅美国医疗保健系统的支出即达每年210亿美元,预计2010年将增长14%,达到240亿美元[6]。年龄增加也是癌症发病的主要危险因素。具体来讲,40-80岁人群的恶性肿瘤的发病率迅速增加,在工业化国家终生癌症风险(lifetime cancer risk)总体为接近1/2[7]。因此,老年性疾病是造成人类不幸的主要原因,消耗了大多数的医疗保健资源。从这个角度看,理解衰老的本质,并设法调控(有可能逆转)衰老的进程,已成为日益迫切的需求。
组织干细胞与增龄性疾病
框1 衰老过程中器官特异性改变 |
衰老过程的解剖和生理变化在发生年龄、进展速度、严重程度方面均存在个体差异,并影响到各种器官和组织类型,涉及高度有丝分裂型,或为静止型。衰老的特征包括肌肉重量减轻(即肌肉衰减症,sarcopenia)、骨骼肌活动能力下降、骨含量降低(即骨质疏松症,osteoporosis),以及皮肤变薄弹性降低(即起皱,wrinkling)。造血系统老化可见进行性正常细胞性贫血,免疫细胞谱变化,如D8+/CD4+T细胞比值增加,骨髓中髓系细胞聚集。其他组织,如脑、肝、肾均可见不同程度的细胞缺失和全重减轻。这种增龄性组织细胞结构变化,与基线生理功能下降密切相关,导致对各类应激的反应程度显著降低,而疾病发生的风险增加。这种功能性降低最突出地表现在肌肉骨骼系统,可见运动水平、强度和动作范围的降低,原因在于肌肉僵硬和关节病变。另外,衰老过程中,伤口愈合和增殖性应激反应均全面退化,如外伤或手术后伤后愈合不良,感染后免疫细胞增殖反应低下。衰老个体还可见肝肾对药物代谢功能降低,以及心率、心肌收缩力和肺活量最高值的降低[91]。 |
人体终生具有非凡的能力,以保持广泛和持续的组织更新。这种持续的自我更新能力源于成体组织干细胞库(reservoir)的维护[8,9]。在衰老和再生研究领域,组织干细胞日益受到重视;越来越多的证据表明,增龄性生理衰退伴发增殖反应削弱(特别是在高度增殖器官),和驻留(resident)组织干细胞异向分化(图1)。同时,上述长寿命可更新的干细胞库中,某些细胞可能有恶化倾向,从而对衰老个体造成负面影响[9]。
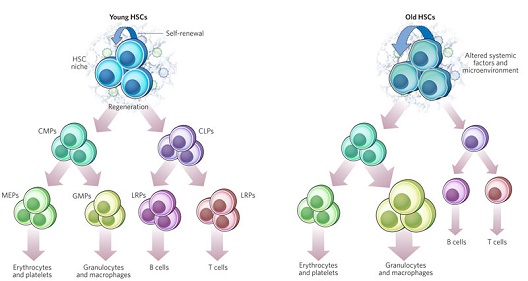
图1 增龄性造血干细胞(HSC)功能下降
图示为年轻(左)和衰老(右)造血干细胞(HSC)的主要差异,提示干细胞增龄性变化的普遍机制,即再生潜能降低和分化失调。细胞内源性和外源性因素共同导致衰老HSC的整体功能衰退。虽然老年HSC的自我更新能力可能会增强,但其再生能力下降,特别是在应激条件下。重要的是,衰老HSC分化程序的改变导致淋巴系祖细胞(CLP)生成减少,而年轻HSC则以相同的速率生成髓系祖细胞(CMP)。较之CLP、成熟B细胞和T细胞数量的减少[称为免疫衰老(immunosenescence)],粒细胞-巨噬细胞系祖细胞(GMP)的生成频率增加,随之粒细胞和巨噬细胞数量增加。而巨核细胞(megakaryocyte)-红细胞系祖细胞(MEP)数量无变化。与HSC功能相关的外源性因素可能包括细胞基质组分和细胞因子谱的改变,从而启动定向分化程序,引发诸如淋巴细胞生成增加,髓细胞生成减少等变化。其他增龄性变化也可能会影响成骨细胞和血管内皮,并显示可调节HSC功能。并生(联体共生,parabiosis)研究显示,干细胞的功能调节与全身因素相关;此项研究将老年小鼠肌卫星细胞暴露于年轻小鼠循环系统,结果显示该细胞再生能力增强(通过Delta–Notch信号的修复效应)[20]。LRP:谱系限制型祖细胞(lineage-restricted progenitors)
各类组织具有不同程度的基础增殖活性和再生潜能。在高度更新(high-turnover)组织,驻留干细胞可产生大量特化后代细胞(specialized cell progeny),以终生维持组织细胞的结构和功能。尽管脑部已发现有干细胞存在[10],但在心脏等增殖能力或再生能力较低的组织,干细胞的鉴定仍颇为困难。凭直觉似乎可提出如下假说,即保持组织干细胞池的充盈及其更新潜能的稳健,对于维持增龄器官功能是至关重要的。支持这一观点是的小鼠早衰模型的表型研究,此类模型小鼠通过条件性删除(conditional deletion)ATR、FOXO转录因子或ATM编码基因获得[ATR:毛细血管扩张性共济失调症及Rad3相关蛋白(ataxia telangiectasia and Rad3 related);ATM:毛细血管扩张性共济失调症突变蛋白(ataxia telangiectasia mutated)];这些小鼠可表现为组织干细胞缺陷,或氧化防御功能减弱及ROS诱导性干细胞损耗(框2)。虽然上述基因模型只具有提示意义,但重要问题是,组织干细胞数量减少或功能降低是否促使老年人健康水平衰退,或至少为某些衰老问题的根本原因。而造血干细胞(HSC)、神经干细胞(NSC)和肌肉干细胞为三类器官系统中已确定的干细胞,且具有不同的增值潜能;为此,针对三者的增龄性分析已证明极具启发性。
造血系统
由于存在细胞自主性及微环境(又称生态位,niche)因素,衰老人群和小鼠的造血系统会出现增龄性干细胞功能降低[9]。而与造血系统变化一致的,则是作为效应器的各血细胞谱系的变化:先天性免疫系统(自然杀伤细胞活性降低,中性粒细胞和巨噬细胞吞噬能力降低,以及促炎症状态)和适应性免疫系统(初始B细胞和T细胞减少,记忆B细胞和T细胞增加)的功能降低,并伴髓系扩增和轻中度正常红细胞性贫血[11]。要特别注意的是,针对骨髓移植患者的回顾性分析显示,供体年龄是显著影响受体存活的唯一参数;现有解释认为年长供体HSC功能会出现细胞自主性的损害[12]。另一条重要证据是,衰老小鼠研究也明确支持上述衰老人群研究,表现在衰老小鼠功能正常的HSC减少,并在细胞自主性作用下倾向于髓系分化,而非淋巴系分化[9]。
框2 干细胞损伤和衰老的遗传模型 |
ATR缺失成年小鼠最突出表现为皮肤、小肠及骨髓中组织特异性干细胞和祖细胞损耗,并见于其他系统,导致广泛的组织萎缩。在数月内,即便有极少的ATR功能完善的干细胞足以恢复组织功能,但小鼠仍出现各种衰老表型;对此可能的解释是再生性反应的强化反可促进早衰,而干细胞微环境的扰动也不能排除[92]。FOXO蛋白(为较少进化生物体的寿命调节因子)可调节氧化防御基因的表达,以消除细胞外ROS毒性[21,22]。生殖细胞系FOXO删除小鼠可见造血干细胞(HSC)和神经干细胞(NSC)的损耗。FOXO缺陷HSC的移植潜能降低,而FOXO缺陷NSC的体外更新潜能和支持脑内神经元新生的功能同样降低[21,22]。与之类似,ATM缺陷小鼠成年期可见进行性骨髓衰竭。在ATM和FOXO突变模型均出现HSC衰竭,这由ROS浓度增加所介导,经抗氧化剂治疗后,可挽救HSC功能和骨髓衰竭[22,93]。 |
神经系统
在人类和小鼠脑部,神经新生(neurogenesis)在整个成年期均持续存在。新生神经元源自NSC,其位于脑室下区和海马齿状回颗粒下层[13]。位于颗粒下层的干细胞后代通过吻侧迁移流(rostral migratory stream)进入嗅球,并分化为中间神经元;而齿状回颗粒下层干细胞后代则迁移到颗粒层,与此处细胞融合[13]。关于增龄性神经新生的变化,大量研究来自于啮齿类;在人类仅少量研究,显示人脑增龄性神经新生减少。老年小鼠(>2岁)的NSC数量较之年轻小鼠减少一半(由各自神经球数量反应)[14,15]。伴随新生成神经元数量呈现增龄性下降,还表现为显著的功能衰退。比如在衰老小鼠,由于脑室下区NSC和神经新生减少,特别是嗅觉上皮细胞持续更新能力降低,导致其嗅觉辨别能力受损[16]。
肌肉系统
骨骼肌再生潜能也表现为增龄性降低,在人类表现为肌纤维生成和纤维组织更新减少;无脊椎动物情形与此类似。[17]。在人类,外伤后再生能力降低是最明显的衰老特征,表现为延迟康复和不完全康复[18]。再生功能损害或可归因于驻留干细胞(卫星细胞,satellite cell)受损。在人类和小鼠衰老过程中,骨骼肌增殖和分化能力降低,并可能伴卫星干细胞数量减少[17,19]。卫星细胞的增龄性变化似主要由“微环境老化”所推动,因为小鼠联体共生实验发现,将老年小鼠肌卫星细胞置于青年小鼠体内,该细胞功能可完全恢复[20]。
衰老的分子通路
针对上述三类典型组织干细胞的机制分析显示,存在数条分子通路与增龄性变化相关联。
首先是PI(3)K通路,该通路可以强烈改变干细胞数量和活性,与衰老和寿命调节密切相关。比如,作为PI(3)K通路下游关键组分的FOXO蛋白破坏,可致ROS诱导性干细胞损耗[21,22]。同样,删除该通路下游另一组分TSC1[mTOR(哺乳类雷帕霉素靶蛋白,mammalian target of rapamycin)负调节因子之一],会导致HSC极度衰竭,并伴ROS浓度增加和异常骨髓动员[23]。TSC1-mTOR信号轴的重要性在药理和基因层次已获证实:在Pten敲除小鼠,HSC表型呈高度增殖状态,并继发骨髓衰竭,而雷帕霉素(一种mTOR抑制剂)则可逆转之;针对mTOR活性下游调节因子——核糖体蛋白S6激酶1(Rps6kb1)的小鼠研究发现,将编码该蛋白的基因删除,可延长小鼠寿命并减轻增龄性病变[25]。
其次是DNA修复途径。具体来讲,DNA修复功能缺陷小鼠可见早衰及神经变性病变,并伴HSC等多种干细胞缺失,而损害DNA修复途径的精确性则注定会发生上述结局。比如核苷酸切除修复(excision repair)缺陷可导致早老综合征(progeroid syndrome)症状,而错配修复(mismatch repair)缺陷则增加癌症风险,但并不加速衰老。端粒功能紊乱所致的DNA损伤亦可表现为早衰和寿命缩短,并致各组织干细胞储备近乎枯竭而见广泛的组织萎缩。上述干细胞缺失的主因在于p53介导的细胞增殖抑制、衰老和/或凋亡消除效应(下文详述)[26,27]。经基因工程使某品系小鼠生殖细胞系等位基因表达高活性p53突变(该基因编码p53蛋白,也称为Trp53),该型小鼠可见早衰表型强化和干细胞功能降低,故可证实衰老与p53激活存在相关性。
第三是细胞死亡通路的分子,如肿瘤抑制因子p16;其表达增加可见于老年人群和小鼠组织(包括小鼠HSC,NSC和胰岛β细胞[30]),似乎与衰老进程相关。而相应地,p16缺失小鼠则显示增龄性HSC、NSC和胰岛β细胞功能增强[30]。
最后是线粒体途径。人们越来越认识到,线粒体功能完整对于干细胞的维护至关重要;已证实在线粒体DNA聚合酶γ基因突变小鼠,由于线粒体DNA突变增加,导致HSC功能受损、严重贫血及淋巴细胞减少[31]。最新证据还表明,BMI1[Ink4a/Arf基因座(也称Cdkn2a)负调节因子之一]缺失,可致线粒体功能降低,表现为电子传递流量降低,引发ROS浓度增加和干细胞受损[32]。而在FOXO缺陷致氧化防御功能缺失的情况下,证实ROS可加速端粒缩短,并激活p53-p21轴,同时调节必要的信号通路以保持HSC和NSC稳定[21,33,34]。
总之,上述不仅有占多数的动物模型研究,也有确凿的人类相关研究,均可证实不同器官的干细胞均存在增龄性增殖和分化潜能降低,而这可能并不反映于干细胞数量的减少。与干细胞功能衰退伴发的是器官功能受损、应激条件下生理反应降低,以及疾病发病率增加。从具体机制来讲,诸多的遗传因子和信号通路通过调节干细胞和线粒体功能,有可能促进衰老进程。尤其值得注意的是,这些遗传因子可通过多种途径,参与基因毒性应激所致的DNA损伤反应(下文详述)。
端粒与衰老
端粒是人类染色体末端起修饰作用的特殊结构。早期研究表明,端粒最重要的作用是保证染色体的完整性[35-37]。而这种核蛋白帽(nucleoproteincap)结构的维持有赖于端粒酶[38-40]。早期的原代人成纤维细胞(fibroblast)研究即推测,充分的端粒酶活性和稳定的端粒长度,对于培养细胞的复制潜能和生物体衰老进程均具有重要意义。在培养细胞,成纤维细胞的分裂伴随端粒逐渐损耗,在经过有限次的细胞分裂后,最终进入增殖停止状态(或称细胞衰老状态),这种限制性称为海弗利克极限(Hayflick limit)[41]。若增殖超越这一极限,将促使端粒进一步损耗,进入染色体断裂-融合-桥(breakage–fusion–bridge)的事件循环,最终引发严重的染色体不稳定状态[42]。
数项突破性进展开创了端粒研究体系。在体外培养的人成纤维细胞中强力表达端粒酶催化亚基——端粒酶逆转录酶(telomerase reverse transcriptase,TERT),细胞可保持端粒长度及无限的复制潜能,而无恶变特性[43,44]。在多种其他类型人类细胞,实验性诱导端粒酶活性可产生强大功效,使细胞能够规避衰老,并实现无限增殖。这些令人信服的细胞培养研究,以及补充性端粒酶基因敲除小鼠研究(见下文),激发研究人员努力探索端粒的动力学机制与衰老进程和/或人类退行性疾病相关性。
人群研究显示,外周血白细胞端粒缩短的60岁以上个体具有较高的死亡率,而最近的一项大型队列研究并未发现上述相关性,但报告端粒长度与健康生活年限存在正相关[45,46]。相对应的一项最近研究发现,百岁老人及其后代的端粒长度与长寿之间亦为正相关;特别是端粒较长者总体健康状况(增龄性疾病少发、认知功能和血脂水平良好)较对照组为佳[47]。进一步强化上述人类研究的证据是,初步研究发现小鼠的正常衰老也与端粒相关(框3)。
框3 野生型小鼠端粒与衰老的相关性 |
处于肿瘤抵抗背景下(该模型小鼠引入了野生型p53和Ink4a/Arf基因位点的额外拷贝)的TERT转基因小鼠,可出现TERT过表达,导致中位寿命(median lifespan)增加,并伴年轻化的特征,如表皮增厚、皮炎减轻、胃炎和肠炎减轻伴体内肠粘膜屏障功能改善,以及离体表皮干细胞克隆活性(clonogenic activity)增强[94]。尽管推测此模型未出现端粒功能紊乱,但有理由认为TERT过表达可稳定表皮干细胞的端粒,进而提示野生型小鼠端粒长度可影响干细胞活性。事实上,近期的野生型小鼠研究汇集的证据(尽管是初步的)支持如下观点,即端粒缩短对于此类模型小鼠寿命有重要作用。上述研究还显示,从短端粒的地中海小家鼠(Mus spretus)到长端粒的小家鼠(Mus musculus)C57BL/6品系,各类小鼠的不同组织均确定存在不同程度的端粒缩短现象[95,96]。C57BL/6小鼠品系在存活至2岁时端粒急剧缩短,并影响到已分化细胞和祖细胞,并导致功能衰退[96]。进一步发现,过表达端粒酶的长端粒小鼠可预防增龄性端粒缩短,延缓表皮和小肠干细胞的功能下降;当然在过表达端粒酶情况下,也可能存在独立于端粒长度的机制[68,94]。最后,研究发现,由于端粒缩短加速,CAST/EiJ小鼠端粒可自然缩短至不足15kb,可表现为单倍体不足(haploinsufficient),或端粒酶完整,最终因高度增殖器官功能退行性降低而亡,涉及病变包括小肠绒毛萎缩、睾丸细胞减少、造血系统全血细胞减少[97,98]。 |
关于端粒在疾病中的作用,一个突出的表现是,端粒长度的保持与心理应激水平及精神疾病风险相平行[48-50]。在20-50岁女性,心理应激水平最高者,其外周血白细胞端粒最短,且端粒酶活性最低,表明该组人群处于最高水平的氧化应激[49]。尤其耐人寻味的是,对于慢性心理应激易感者寿命较短,典型增龄性疾病(如心血管疾病、免疫功能低下)发病较早[50]。而自主神经系统和神经内分泌系统激活,促使糖皮质激素诱导ROS增加,可能是造成端粒损耗加速和端粒直接损伤的根本原因[51,52]。进一步的推论认为,受损端粒无法有效修复的原因,可能在于端粒酶活性较低,且端粒固有的屏蔽效应可阻止DNA修复机制。因此,受损端粒可能成为持续性DNA损伤信号的会合点,由此继发p53持续激活和衰老后果。
框4 早老综合征(progeroid syndrome)与端粒功能紊乱 |
多种单基因突变显示可诱导不同程度的早衰。根据脏器受累的范围,可将这些遗传性综合征分为两类,累及多脏器和组织者称为节段型早老综合征(segmental progeroid syndromes),以单一脏器为主者称为单峰型早老综合征(unimodal progeroid syndromes)。前者包括沃纳综合征(Werner syndrome)、毛细血管扩张性共济失调症(ataxia telangiectasia)、先天性角化不良(dyskeratosis congenita)、哈钦森-吉尔福特综合征(Hutchinson–Gilford progeria syndrome)以及布鲁姆综合征(Bloom syndrome);后者包括家族性阿尔茨海默病(familial Alzheimer's disease)、家族性帕金森病(familial Parkinson's disease)以及衰减型家族性息肉病(attenuated familial polyposis)[99]。 沃纳综合征为罕见的常染色体隐性遗传疾病,每10万人中有1人发病,病因为WRN蛋白缺陷(WRN蛋白为RecQ DNA解旋酶,参与DNA修复、DNA重组和端粒维护)。在沃纳综合征细胞可见染色体高度重组且多重突变,提示由于异常重组事件无法抑制和基因组整体不稳定性,导致本病衰老加速和癌症易感性增加。重要的是,沃纳综合征患者成纤维细胞显示端粒损耗加速和早衰,可通过加强TERT表达挽救之。本病患者青春期前发育正常,青春期则生长停滞,并开始出现多种进行性早老病变,包括老年性白内障、骨质疏松症、皮肤萎缩、头发花白、心肌梗死和恶性肿瘤。本病可显著缩短预期寿命(中位寿命47-48岁),主要死于心肌梗死和恶性肿瘤[99]。毛细血管扩张性共济失调症也为常染色体隐性遗传疾病,表现为进行性小脑变性、皮肤异常(毛细血管扩张、皮肤萎缩、色素异常和头发花白)、免疫缺陷及多种恶性肿瘤。本病源自ATM基因的各种致病性突变(ATM蛋白对于DNA损伤信号、DNA修复和端粒维护具有关键作用)。本病患者细胞端粒显著损耗,导致染色体不稳定性加重和早衰,也可通过加强TERT表达挽救之[99]。 引人注目的是,Wrn和Atm缺陷小鼠模型并未能显示人类疾病的典型变性病变。然而,若将上述小鼠置于端粒酶缺陷的背景下(伴有限端粒),则小鼠可见多种增龄性变性病变。这些研究观察强调了如下事实,即有限的端粒是显著的限速性(rate-limiting)发病因素,可控制上述遗传性疾病增龄性表型的发生。 |
针对一系列的遗传性退行性疾病的研究推测,端粒对保持健康人群寿命具有重要意义。例如,先天性角化不良症(dyskeratosis congenita)为常染色体显性遗传,目前已知该病患者携带TERT(端粒酶催化成分)或TERC(telomerase RNA component,即端粒酶RNA组分,负责编码RNA模板)突变[53]。这些患者的端粒和寿命均缩短,且具有衰老加速征象,伴发的骨髓衰竭可增加致命性感染风险[53]。针对某些早老病症如沃纳综合征(Werner syndrome)和毛细血管扩张性共济失调症(ataxia telangiectasia),通过对患者及其衍生细胞进行分析,有证据同样显示端粒与衰老进程相关(框4)。然而,TERC或TRET突变更常见于器官特异性功能紊乱,如特发性肺纤维化(idiopathic pulmonary fibrosis,一种致命的进行性肺病,伴肺泡疤痕形成)[54]、骨髓衰竭综合征(bone marrowfailure syndromes)[55];以上强烈提示端粒长度维持机制的缺陷,造成了这些病症复杂的组织特异性表现。除了遗传性疾病,端粒似乎也与获得性退行性疾病(表现为组织更新速度逐渐增加)相关。最突出的实例是肝硬化,位列全球最常见致死性疾病第七位[56];该病表现为端粒储备进行性下降,而肝细胞更新加快,导致肝细胞持续增殖中止并凋亡,最终肝功能衰竭。
综上所述,各类人类退行性疾病(包括遗传性或获得性)的研究证据均指向了端粒,其作为关键性的发病因素促成退行性病变、增加癌症风险,并且缩短寿命。就这一点而言,端粒储备容量、持续性损害(或有益因素)水平,以及端粒帽状态测定值,均可能成为疾病进展的生物标志物,并可能提供新的契机以实现积极主动的干预治疗(如通过短暂性激活体细胞内源性端粒酶以补充或修复端粒)。
端粒酶基因敲除小鼠
在早期,通过Terc或Tert基因敲除小鼠的制备和鉴定,从在体角度确定了端粒与衰老、退行性疾病和癌症发生的相关性及具有的特定功能。值得注意的是,Terc或Tert基因敲除小鼠的健康状况和表型似乎未受影响,似可确定端粒酶活性对小鼠生存并无大碍[57,58]。而第一代(G1)Terc−/−小鼠表型显著缺乏的原因,据推测可能由于端粒储备尚足以维持端粒帽的功能57。事实上,Terc−/−小鼠后代则表现为端粒的极度缩短和染色体末端-末端融合(end-to-end fusions)。与端粒功能紊乱[无信号末端(signal-freeends)和末端-末端融合]细胞遗传学证据相一致的是,后代(G3及以后)的Terc-/-小鼠的寿命缩短、整体功能衰退、繁殖能力下降、组织萎缩伴器官功能受损[59](图2)。广泛的组织退行性病变表型包括贫血及淋巴细胞减少、脊柱后凸(kyphosis)及骨质疏松(osteoporosis)、轻度糖耐量受损,以及其他典型增龄性表现[26]。Terc-/-后代小鼠的上述退行性表型的严重程度与端粒功能紊乱程度相平行;端粒功能可通过测定染色体融合和分裂后期桥体(anaphasebridges)的数目加以判断,而最近研究则是通过测定端粒部位DNA损伤位点数目实现[60,61]。此外,在表达典型的人类退行性疾病突变(如WRN和ATM)的基因工程小鼠,只有在端粒酶无缺陷、也无端粒缩短和功能受限情况下,才突出表现为衰老表型延迟[62,63],进一步强化了端粒与衰老进程存在相关性的观点。
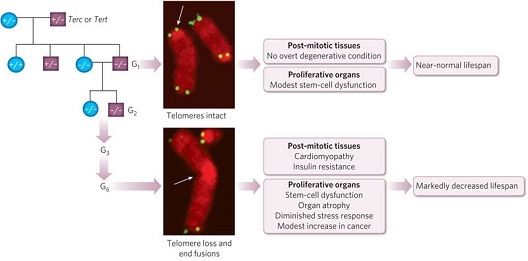
图2 端粒酶基因敲除小鼠端粒功能紊乱并伴发早衰
端粒酶基因敲除小鼠(G1)可完全存活,染色体完整,并拥有长端粒(上部图片:箭头处),因此较少有生理异常表现;而在衰老过程中,G1小鼠退行性症状的发生要早于同龄Terc野生型小鼠。将端粒酶基因敲除小鼠连续种间杂交产生的后代小鼠(G2、G3,……),则见端粒长度缩短,端粒功能紊乱小鼠可见染色体形态异常(下部图片:箭头指向示端粒信号缺失,导致染色体融合),而这些小鼠衰老过程中则会出现多种退行性病症,并见于高度增殖器官和有丝分裂后组织。高度增殖器官(如肠道、皮肤和睾丸)典型特征为萎缩性变化,提示为干细胞功能的衰竭;而有丝分裂后组织的功能衰退(如心肌病)及增龄性代谢异常(如胰岛素抵抗),在端粒功能紊乱小鼠也有发现。这种小鼠的寿命缩短,且癌症发生有所增加,与端粒防止异常重组(illegitimaterecombination)事件的功能相呼应。
造成上述退行性表型与端粒之间显著相关性的基本机制是什么?对高度增殖器官来讲,其有赖于通过驻留的组织干细胞分化以实现器官更新和稳态;且有明确证据显示,在Terc-/-小鼠后代的各类组织中,存在干细胞和祖细胞的储备枯竭和/或功能受损;具体经由不同途径所导致,如细胞凋亡增加、衰老以及分化受损。例如,衰老的Terc-/-小鼠后代小肠萎缩程度与高水平的p53依赖性凋亡相平行,此类型凋亡见于干细胞和祖细胞丰富的小肠隐窝区(cryptcompartment)[26]。此外,Terc-/-小鼠后代HSC在竞争性移植(competitivetransplantation)实验处于严重弱势,并显示明显向髓系分化,而这一表现与衰老人群类似[64,65]。皮肤研究发现,Terc-/-小鼠后代表皮干细胞增殖能力及移出毛囊的能力均受损,而这些缺陷可能造成伤口愈合延迟,毛发灰白及脱发[66]。在脑部,虽然其增殖活性相对较低,Terc-/-小鼠后代NSC仍显示为更新和分化能力受损[63,67]。部分分子通路可感受端粒功能紊乱并激活细胞检测点反应,进而导致干细胞缺陷和和组织退行性表型;虽然对各通路仍不完全了解,但p53已显示具有显著的作用(见下文)。
端粒酶与干细胞稳态
欲了解端粒酶在退行性疾病和癌症中的作用,目前研究多源于端粒酶基因敲除小鼠,且聚焦于端粒帽的功能;而在干细胞生物学研究中有关端粒酶功能的新证据,已不再涉及其端粒维持功能。在某项转基因系列研究中,强化TERT在皮肤中的表达,结果显示可激活静态毛囊干细胞,刺激毛发生长。这种TERT诱导的干细胞激活效应与TERT的经典的端粒合成功能并无关联,因为在端粒酶缺陷(Terc-/-)或酶失活型TERT转基因背景下,TERT的这一功能同样存在68 。这些研究指出TERT的端粒非依赖性功能在组织干细胞稳态中亦可实现,而由于人类TERT可与内切核糖核酸酶(endoribonuclease,一种核糖核蛋白,即RNase MRP复合体)的RNA组分(RMRP)相结合[69],TERT在此领域的研究受到高度重视。从我们研究模型的关联性判断,此部分的报道将引发关注,这是由于RNaseMRP复合体涉及广泛的细胞和线粒体功能;并且RMRP突变患者可表现软骨-毛发发育不全综合征(cartilage–hairhypoplasia syndrome),以多器官功能衰竭为特征(主要见于高度增殖器官,与可能的干细胞衰竭相一致)。因此,针对TERT越来越多的关注已经超越其端粒维持功能,而在于其可能与线粒体和干细胞的功能优化相关,进而影响到增龄性进程。
p53肿瘤抑制因子与基因组维护
通过对DNA修复机制缺陷患者的遗传学研究,可确定衰老过程中保持基因组完整性具有重要意义。小鼠遗传学研究也强化了DNA修复过程在衰老和退行性疾病中的重要作用。在应对DNA损伤及继发产生退行性衰老表型的过程中,p53信号发挥了重要作用,表现在其可通过删除作用挽救(实际是几乎完全逆转)上述表型。
基因组维护
伴基因组维护缺陷的人类综合征往往与衰老加速相关。伴类似突变的基因工程小鼠显示,完整的DNA修复机制在干细胞维护和衰老进程方面发挥了重要作用。在Ku80(也称XRCC5)缺陷或XPD(也称ERCC2)缺陷的年轻小鼠,其HSC未见明显的DNA损伤或功能衰退;而相应的衰老小鼠HSC则显示DNA损伤位点聚集,在移植条件下发现小鼠再生能力受到显著影响[64]。尽管上述实验结果与正常衰老的相关性并不明确,但值得注意的是,在老年野生型小鼠HSC可见DNA损害位点数量增加,且功能损害程度加重[64]。在另一种DNA修复机制缺陷基因工程小鼠,涉及到连接酶IV的亚效等位基因(hypomorphicallele),还有一种为MSH2和Fancd1(也称BRCA2)敲除小鼠,均显示DNA损伤增加和干细胞数量减少[8]。针对具有Rad50超效等位基因(hypermorphic allele)的小鼠,在无过度DNA损伤情况下,超激活(hyperactivation)其DNA损伤反应通路,也可诱导干细胞损耗;且小鼠干细胞凋亡增强导致骨髓衰竭,因此其寿命仅为四周[70]。最近研究表明,电离辐射所致DNA损伤可导致黑色素细胞的干细胞损耗,这种细胞类型的损耗机制在于其可迅速退出静止态并加强分化,而非经由凋亡和衰老途径[71]。此外,值得注意的是,某些DNA修复蛋白(包括RAD50、Ku70或Ku80、ATM和WRN),均对于端粒的维护至关重要。另一个需重视的蛋白是SIRT6,为sirtuin家族成员且与端粒相关,对于DNA稳定和修复相当重要。删除SIRT6编码基因可诱导端粒功能紊乱伴染色体末端-末端融合,导致衰老细胞增加,衰老表型明显加速,预期寿命仅为3周[72,73]。总之,上述DNA修复机制缺陷基因工程小鼠模型提供了确凿的遗传学证据,表明基因组完整性维持与机体衰老进程之间存在相关性。
p53肿瘤抑制因子
作为基因组“监护者”(guardian),p53是重要的细胞应激感受器;在端粒功能紊乱等DNA损伤以及ROS、癌基因激活和缺氧等不良刺激情况下,p53可被激活从而作出反应。p53激活可致细胞生长停滞并修复,或发生细胞凋亡和衰老;而细胞的不同结局取决于p53的激活程度[74]。生殖细胞系p53删除小鼠的端粒则极度缩短,为保持p53相应功能,该小鼠可见细胞凋亡显著降低,而各组织的增殖水平显著提高[27]。在小鼠p53缺陷背景下,如逆转干细胞损耗,可改善睾丸、肠和皮肤等器官以及造血系统的功能。Terc-/-p53-/-小鼠后代的伤口愈合、毛发生长及皮肤更新情况均优于野生型小鼠,并见其表皮干细胞数量增多及动员能力提高[75]。同样,Terc-/-p53-/-小鼠后代HSC移植研究亦显示HSC增殖能力得以提高[64]。总的来说,以上资料表明,p53依赖性端粒检测点机制在各类型干细胞中均发挥作用。
虽然p53删除小鼠的干细胞功能改善,但端粒缩短且功能紊乱,而由于癌症发病率的增加,小鼠的整体寿命并未延长。相对于Terc-/-p53+/+小鼠后代,Terc-/-p53-/-(或p53+/-)小鼠后代的肿瘤发病率增加,重要的是肿瘤谱也发生变化[76]。p53-/-小鼠典型表现为淋巴瘤和肉瘤为主的肿瘤谱,且端粒完整;而Terc-/-p53-/-小鼠的端粒功能紊乱,并出现皮肤、胃肠道和乳腺肿瘤,而这些肿瘤与老年人群的肿瘤谱高度相关[76]。p53缺陷小鼠的端粒功能紊乱确可影响衰老和癌症表型,还可将Ink4a/Arf删除小鼠与其作比较;Ink4a/Arf编码p16 INKA4(细胞周期蛋白依赖性激酶抑制剂)和p19ARF(p53激活因子)[30]。Ink4a/Arf删除小鼠表现为端粒功能紊乱,退行性表型因此未能改善,且上皮细胞肿瘤发病率增加。而与Ink4a/Arf-/-小鼠的发病预期相比,Terc-/-Ink4a/Arf-/-小鼠后代罹患淋巴瘤和肉瘤致死,但潜伏期较长[77]。以上对比研究强调,遗传背景可通过端粒在肿瘤和衰老进程中发挥显著作用,同时也凸显了p53的重要性;反之,由于Ink4a/Arf缺陷小鼠无法实现DNA损伤诱导信号的保存和p53激活,该型小鼠缺乏p53补救功能[77]。事实上,Terc-/-Ink4a/Arf-/-小鼠后代可见p53激活伴凋亡增加,并显示睾丸、小肠萎缩,以及造血功能受损[77]。是否这些缺陷与组织干细胞枯竭存在明确相关性,尚需深入研究。
Ink4a/Arf缺陷小鼠因端粒功能紊乱,其退行性表型完全不能改善,这一改变与报道的Bmi1-Ink4a/Arf轴在干细胞维护中的作用并不一致[30];Ink4a/Arf缺陷小鼠可补救Bmi1缺陷所致的HSC-NSC损耗表型,而通过Ink4a定向突变(targetedmutation)可致p16 Ink4a的特异性缺失,增强衰老HSC、NSC、淋巴细胞和胰岛β细胞的再生潜能30。也就是说,Bmi1缺陷所见的干细胞损耗表型,不仅与Ink4a/Arf[BMI1是Ink4a/Arf位点(locus)的阻遏蛋白(repressor)]表达上调相关,而且还与的线粒体功能紊乱加重相关,从而导致ROS浓度增加并继发DNA损伤反应通路的激活[32]。因此容易推测Bmi1缺陷背景下ROS浓度升高,可造成端粒的损伤和消耗,并继发p53激活增强;而在Terc-/-Ink4a/Arf-/-小鼠后代,Ink4a/Arf缺陷即便对干细胞和祖细胞具有改善作用,也会被p53激活增强效应所完全消除。Bmi1-Ink4a/Arf调节线粒体的生物学机制仍然是需要继续研究的重要领域。
两种p53突变遗传工程小鼠进一步证实了p53激活在机体衰老中的作用:这些小鼠出现早衰,且具显著肿瘤抗性[28,29]。衰老过程中,这些超级p53突变小鼠的不同组织均表现为衰老细胞数量增加;而且,竞争性移植发现,其HSC活性降低和移植容量(engraftmentcapacity)均降低[78]。而在Terc、Brca1、Zmpste24(人类哈钦森-
|
图3 DNA损伤、p53激活和线粒体功能紊乱交互作用模型 |
在此模型中,引发基因毒性应激因素有端粒损耗、DNA修复受损、紫外线(UV)辐射、电离辐射(IR)、化学品、ROS及其他机制,均可激活p53并诱导细胞生长停滞(在增殖部位)、衰老或凋亡。我们还提出,p53可直接或间接损伤线粒体功能(通过ROS解毒酶的调节)。p53介导的线粒体功能紊乱可引发DNA损伤、p53激活、线粒体受损、ROS增多,再DNA损伤的循环。线粒体受损所致的ATP生成减少,再加上线粒体的代谢改变,均促使器官功能紊乱。p53与其他衰老相关通路之间也提示存在相互作用。热量限制(CR)可激活SIRT1,后者可降低p53活性。此外,SIRT1(SIRT6可能)可激活PGC-1α,并加强线粒体的生物合成[2]。PGC-1α可通过上调抗氧化剂增强氧化防御功能[89],且已证实p53可根据细胞ROS浓度变化,增减抗氧化剂的生成[86]。BMI1缺失在直接诱导线粒体功能紊乱的同时还诱导p16/ARF上调[30,32]。ARF可通过与MDM2(p53的负调节因子)相互作用,增强p53的活性。 |

吉尔福特早老综合征的小鼠模型)或Ku80等基因删除小鼠[79,80](其他早老综合征见框4),由于p53缺陷导致无法补救多种衰老加速表型,这与前述p53突变小鼠所见相一致。而额外携带野生型p53和/或Ink4a/Arf位点完整拷贝的转基因小鼠,其寿命正常或延长达16%以上,同时衰老表型延迟出现,这可能与ROS浓度较低,以及ROS相关性蛋白和脂质损害减轻有关[81]。然而p53信号仍具有相当的复杂性,具有Mdm2(p53主要负调节因子)亚效等位基因的小鼠,在p53活性增强的情况下,未见明显的早衰迹象[82]。最后,在端粒功能极度紊乱和染色体极不稳定的背景下,失活p53可能并不足以维持生存能力[ 83,84]。
p53功能迥异的两个方面(促进衰老和延缓衰老)可能并不存在排他性(non-exclusive),而与其活性水平、激活动力学(kinetics)及所处细胞或遗传背景相关。虽然p53删除小鼠可见细胞和机体衰老表型的逆转,但是p53失活针对衰老和干细胞的益处,却因同时增加癌症风险而被抵消。上述结果强烈提示尚需深入揭示p53介导的检测点网络环路,如此方有可能发现延缓衰老进展且抑制肿瘤发生的途径。沿着这一思路,值得注意的是,在Terc-/-小鼠后代,同时删除p21Cip(为p53靶点和细胞周期负调节因子),可减轻多个器官的组织变性,且不增加肿瘤风险[65]。因此,进一步系统解析p53网络,将可能为衰老和癌症风险的优化管理,展示新的治疗手段。
展望
表面看来,基因毒性应激(特别是受损端粒所致)似乎是造成相对静态器官(如心、肝)增龄性功能衰退的基础,但两者间较少存在相关性。然而,心肌病是却正是的Terc-/-小鼠模型后代的突出表型[85]。因此,我们提出一种模型,来描述存在于这些高代谢率器官的相关机制:即线粒体功能紊乱是导致基因毒性损害循环逐步加重的基础,该循环依次表现为p53激活、线粒体功能紊乱、ROS浓度增加,再进一步损伤DNA[2]。特别是线粒体储备减少和功能降低可见于老年人群和小鼠组织,由此导致ROS生成增加,而这主要归因于复合体Ⅰ和复合体Ⅲ的功能紊乱。如ROS生成大幅增加将启动如下的有害循环,即基因毒性损害增加造成端粒迅速损耗,再继发持续的p53激活,线粒体功能进一步衰退,又会产生更多的ROS,如此循环往复[34]。这种恶性循环也可能解释晚年衰老症状的累积性(cumulativeand precipitative nature)。
我们注意到,在上述损伤-反应循环过程中,p53激活所致的细胞反应可能与ROS浓度阈值相关。低氧化应激情况下,p53激活优先诱导抗氧化剂基因表达;而当ROS生成增加时,p53却激活促氧化剂(pro-oxidant)基因[86]。这种p53的反差表现也可见于下列情况:在DNA适度损伤情况下,细胞周期停滞并修复;而在DNA损伤持续存在情况下,可发生更强烈的细胞反应,细胞发生衰老、凋亡和/或线粒体功能紊乱,从而导致组织萎缩和功能衰退[74]。
如下基因毒性应激衰老模型(图3)以端粒-p53轴为核心,其与衰老过程中几乎所有的重要遗传成分均能实现良好结合。首先,该轴可解释端粒功能紊乱小鼠和生殖细胞系p53超激活小鼠并见早衰表型的原因[28,29]。其次,该轴可解释缺乏SIRT1或SIRT6(均为降低p53活性的蛋白)小鼠发生早衰的原因[87]。第三,该轴可以解释线粒体与关键衰老因子(如PGC-1α、PGC-1β、FOXO蛋白及BMI1)之间的联系;小鼠若缺乏编码这些蛋白的基因,则发生组织变性加速及线粒体功能紊乱。
以“衰老轴”为核心,线粒体与之融为一体,这一观点得到了各种早衰模型的支持(如端粒功能紊乱小鼠、超级p53小鼠);还有某些小鼠具有过量的线粒体DNA突变,或缺乏PGC-1α、PGC-1β(两者均为线粒体生物合成和代谢的主调节因子)[88-90]。然而,就上述早老表型的共性因素而言,其精确的分子机制仍有待揭示。在端粒酶基因敲除小鼠,其大量的有丝分裂后器官可见明显的功能衰退;受此启发,我们推测p53激活可能会通过某种未知机制,导致线粒体生物合成和/或其功能衰退。一旦连接p53与线粒体功能紊乱的特异性分子组成得以确定,将使衰老中心轴的基础相统一。将遗传毒性应激与干细胞损伤,线粒体功能衰退,及最终的器官萎缩和功能衰退、能量生成减少相联系,将体现衰老机体细胞和生理各方面衰退的本质特征。事实上,当我们考虑“普遍性衰退”(generalizedfrailty)这一衰老的标志性特征时,会联想到这源于基础性功能紊乱所致的细胞ATP生成不足,那么本文对线粒体研究的展望必将受到极大的关注。提出这个遗传毒性应激衰老模型后,有必要进一步理解影响端粒损耗的因子、保护细胞免受ROS损伤的基因、调节p53活性的信号,以及保持线粒体储备和功能的通路等。还需明确在衰老过程中上述不同途径如何实现各司其职和相互协调。这一网络的破解可能会催生衰老生物标志物和领先的治疗策略[包括通过短暂性再激活端粒酶以稳定端粒,调控p53、改善线粒体功能及生物合成、调控mTOR和PI(3)K通路],实现衰老个体增殖组织和静态组织的全面复原。
参考文献
1. Kenyon, C. Theplasticity of aging: insights from long-lived mutants. Cell 120, 449–460 (2005).
2. Guarente, L.Mitochondria — a nexus for aging, calorie restriction, and sirtuins? Cell132, 171–176 (2008).
3. Finkel, T., Serrano,M. & Blasco, M. A. The common biology of cancer and ageing. Nature 448, 767–774 (2007).
4. Campisi, J. Senescentcells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell120, 513–522 (2005).
5. Kirkwood, T.B. Understanding the odd science of aging. Cell 120, 437–447 (2005).
6. Arrighi, H. M.,McLaughlin, T. & Leibman, C. Prevalence and impact of dementia-related functionallimitations in the United States, 2001 to 2005. Alzheimer Dis. Assoc. Disord.doi:10.1097/WAD.0b013e3181a1a87d (in the press).
7. DePinho, R. A.The age of cancer. Nature 408,248–254 (2000).
8. Sharpless, N.E. & DePinho, R. A. How stem cells age and why this makes us grow old. NatureRev. Mol. Cell Biol. 8, 703–713(2007).
9. Rossi, D. J.,Jamieson, C. H. & Weissman, I. L. Stems cells and the pathways to aging andcancer. Cell 132, 681–696(2008).
10. Zhao, C., Deng,W. & Gage, F. H. Mechanisms and functional implications of adult neurogenesis.Cell 132, 645–660 (2008).
11. Linton, P. J.& Dorshkind, K. Age-related changes in lymphocyte development and function.Nature Immunol. 5, 133–139(2004).
12. Kollman, C. etal. Donor characteristics as risk factors in recipients after transplantationof bone marrow from unrelated donors: the effect of donor age. Blood 98, 2043–2051 (2001).
13. Gage, F. H. Mammalianneural stem cells. Science 287,1433–1438 (2000).
14. Maslov, A. Y.,Barone, T. A., Plunkett, R. J. & Pruitt, S. C. Neural stem cell detection, characterization,and age-related changes in the subventricular zone of mice. J. Neurosci.24, 1726–1733 (2004).
15. Molofsky, A.V. et al. Increasing p16INK4a expression decreases forebrain progenitorsand neurogenesis during ageing. Nature 443, 448–452 (2006).
16. Enwere, E. etal. Aging results in reduced epidermal growth factor receptor signaling, diminishedolfactory neurogenesis, and deficits in fine olfactory discrimination. J. Neurosci.24, 8354–8365 (2004).
17. Cerletti, M.,Shadrach, J. L., Jurga, S., Sherwood, R. & Wagers, A. J. Regulation and functionof skeletal muscle stem cells. Cold Spring Harb. Symp. Quant. Biol. 73, 317–322 (2008).
18. Di Iorio, A.et al. Sarcopenia: age-related skeletal muscle changes from determinantsto physical disability. Int. J. Immunopathol. Pharmacol. 19, 703–719 (2006).
19. Gopinath, S.D. & Rando, T. A. Aging of the skeletal muscle stem cell niche. Aging Cell7, 590–598 (2008).
20. Conboy, I. M.et al. Rejuvenation of aged progenitor cells by exposure to a young systemicenvironment. Nature 433, 760–764(2005).
21. Paik, J. H. etal. FoxOs cooperatively regulate diverse pathways governing neural stem cellhomeostasis. Cell Stem Cell 5,540–553 (2009).
22. Tothova, Z. &Gilliland, D. G. FoxO transcription factors and stem cell homeostasis: insightsfrom the hematopoietic system. Cell Stem Cell 1, 140–152 (2007).
23. Gan, B. etal. mTORC1-dependent and-independent regulation of stem cell renewal, differentiation,and mobilization. Proc. Natl Acad. Sci. USA 105, 19384–19389 (2008).
24. Yilmaz, O. H.et al. Pten dependence distinguishes haematopoietic stem cells fromleukaemia-initiating cells. Nature 441, 475–482 (2006).
25. Selman, C. etal. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science326, 140–144 (2009).
26. Rudolph, K. L.et al. Longevity, stress response, and cancer in aging telomerase-deficientmice. Cell 96, 701–712 (1999).
本文报道在端粒酶敲除小鼠中极短端粒对寿命和应激反应造成的影响。短端粒小鼠可见寿命缩短,应激状态下增殖能力受损,如伤口愈合异常、造血干细胞损耗。
27. Chin, L. etal. p53 deficiency rescues the adverse effects of telomere loss and cooperateswith telomere dysfunction to accelerate carcinogenesis. Cell 97, 527–538 (1999).
本文显示在正常和肿瘤细胞,p53均可介导端粒功能紊乱引发的细胞反应;且p53缺陷可减轻组织变性表型。
28. Maier, B. etal. Modulation of mammalian life span by the short isoform of p53. GenesDev. 18, 306–319 (2004).
29. Tyner, S. D.et al. p53 mutant mice that display early ageing-associated phenotypes. Nature415, 45–53 (2002).
文献28、29报道超激活p53小鼠可见促衰老表型。
30. Kim, W. Y. &Sharpless, N. E. The regulation of INK4/ARF in cancer and aging. Cell127, 265–275 (2006).
31. Chen, M. L. etal. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arisingfrom mitochondrial dysfunction. Blood 114, 4045–4053 (2009).
32. Liu, J. etal. Bmi1 regulates mitochondrial function and the DNA damage response pathway.Nature 459, 387–392 (2009).
本文报道BMI1缺陷小鼠线粒体功能紊乱可增加ROS水平,激活DNA损伤反应;以上可经抗氧化剂治疗而部分逆转。
33. Tothova, Z. etal. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologicoxidative stress. Cell 128,325–339 (2007).
34. Passos, J. F.,Saretzki, G. & von Zglinicki, T. DNA damage in telomeres and mitochondria duringcellular senescence: is there a connection? Nucleic Acids Res. 35, 7505–7513 (2007).
35. McClintock, B.The behavior in successive nuclear divisions of a chromosome broken at meiosis.Proc. Natl Acad. Sci. USA 25,405–416 (1939).
36. Szostak, J. W.& Blackburn, E. H. Cloning yeast telomeres on linear plasmid vectors. Cell29, 245–255 (1982).
37. Shampay, J.,Szostak, J. W. & Blackburn, E. H. DNA sequences of telomeres maintained in yeast.Nature 310, 154–157 (1984).
38. Blackburn, E.H. Switching and signaling at the telomere. Cell 106, 661–673 (2001).
39. Greider, C. W.& Blackburn, E. H. Identification of a specific telomere terminal transferaseactivity in Tetrahymena extracts. Cell 43, 405–413 (1985).
这篇开创性研究发现,四膜虫(Tetrahymena)细胞抽提液中存在端粒酶活性,能够延伸(TTGGGG)n寡核苷酸。
40. Greider, C. W.& Blackburn, E. H. The telomere terminal transferase of Tetrahymena isa ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51, 887–898 (1987).
41. Hayflick, L.& Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp.Cell Res. 25, 585–621 (1961).
42. Maser, R. S.& DePinho, R. A. Connecting chromosomes, crisis, and cancer. Science297, 565–569 (2002).
43. Bodnar, A. G.et al. Extension of life-span by introduction of telomerase into normal humancells. Science 279, 349–352(1998).
这篇开创性研究显示,将端粒酶重新引入人类视网膜上皮细胞和成纤维细胞,可防止细胞衰老,并致人类细胞永生化。
44. Counter, C. M.et al. Dissociation among in vitro telomerase activity, telomere maintenance,and cellular immortalization. Proc. Natl Acad. Sci. USA 95, 14723–14728 (1998).
45. Cawthon, R. M.,Smith, K. R., O'Brien, E., Sivatchenko, A. & Kerber, R. A. Association betweentelomere length in blood and mortality in people aged 60 years or older. Lancet361, 393–395 (2003).
46. Njajou, O. T.et al. Association between telomere length, specific causes of death, andyears of healthy life in health, aging, and body composition, a population-basedcohort study. J. Gerontol. A 64,860–864 (2009).
47. Atzmon, G. etal. Genetic variation in human telomerase is associated with telomere lengthin Ashkenazi centenarians. Proc. Natl Acad. Sci. USA 107 (suppl. 1), 1710–1717 (2010).
48. Epel, E. S. etal. Accelerated telomere shortening in response to life stress. Proc. NatlAcad. Sci. USA 101, 17312–17315(2004).
49. Epel, E. S. etal. Cell aging in relation to stress arousal and cardiovascular disease riskfactors. Psychoneuroendocrinology 31, 277–287 (2006).
50. Simon, N. M.et al. Telomere shortening and mood disorders: preliminary support for achronic stress model of accelerated aging. Biol. Psychiatry 60, 432–435 (2006).
51. Passos, J. F.& von Zglinicki, T. Mitochondria, telomeres and cell senescence. Exp. Gerontol.40, 466–472 (2005).
52. Oexle, K. &Zwirner, A. Advanced telomere shortening in respiratory chain disorders. Hum.Mol. Genet. 6, 905–908 (1997).
53. Kirwan, M. &Dokal, I. Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys.Acta 1792, 371–379 (2009).
54. Armanios, M.Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis.N. Engl. J. Med. 356, 1317–1326(2007).
55. Yamaguchi, H.et al. Mutations in TERT, the gene for telomerase reverse transcriptase,in aplastic anemia. N. Engl. J. Med. 352, 1413–1424 (2005).
56. Rudolph, K. L.,Chang, S., Millard, M., Schreiber-Agus, N. & DePinho, R. A. Inhibition of experimentalliver cirrhosis in mice by telomerase gene delivery. Science 287, 1253–1258 (2000).
57. Blasco, M. A.et al. Telomere shortening and tumor formation by mouse cells lacking telomeraseRNA. Cell 91, 25–34 (1997).
本文承接McClintock假说,通过端粒酶敲除实验手段,提出端粒酶活性对维持端粒稳定和防止染色体末段-末端重组具有至关重要的作用。
58. Farazi, P. A.,Glickman, J., Horner, J. & Depinho, R. A. Cooperative interactions of p53 mutation,telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression.Cancer Res. 66, 4766–4773(2006).
59. Lee, H. W. etal. Essential role of mouse telomerase in highly proliferative organs. Nature392, 569–574 (1998).
60. Takai, H., Smogorzewska,A. & de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol.13, 1549–1556 (2003).
61. Hande, M. P.,Samper, E., Lansdorp, P. & Blasco, M. A. Telomere length dynamics and chromosomalinstability in cells derived from telomerase null mice. J. Cell Biol. 144, 589–601 (1999).
62. Chang, S. etal. Essential role of limiting telomeres in the pathogenesis of Werner syndrome.Nature Genet. 36, 877–882(2004).
63. Wong, K. K. etal. Telomere dysfunction and Atm deficiency compromises organ homeostasis andaccelerates ageing. Nature 421,643–648 (2003).
本文显示ATM缺陷和端粒功能紊乱可共同损伤干细胞和祖细胞储备,对细胞和机体生存造成负面影响。
64. Rossi, D. J.et al. Deficiencies in DNA damage repair limit the function of haematopoieticstem cells with age. Nature 447,725–729 (2007).
本文显示,HSC中DNA损伤的聚集可损害其再生能力,提供了干细胞功能增龄性降低与DNA损害存在相关性的证据。
65. Choudhury, A.R. et al. Cdkn1a deletion improves stem cell function and lifespanof mice with dysfunctional telomeres without accelerating cancer formation. NatureGenet. 39, 99–105 (2007).
66. Flores, I., Cayuela,M. L. & Blasco, M. A. Effects of telomerase and telomere length on epidermalstem cell behavior. Science 309,1253–1256 (2005).
67. Ferron, S. etal. Telomere shortening and chromosomal instability abrogates proliferationof adult but not embryonic neural stem cells. Development 131, 4059–4070 (2004).
68. Sarin, K. Y.et al. Conditional telomerase induction causes proliferation of hair folliclestem cells. Nature 436, 1048–1052(2005).
69. Maida, Y. etal. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA.Nature 461, 230–235 (2009).
70. Morales, M. etal. The Rad50S allele promotes ATM-dependent DNA damage responses andsuppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor.Genes Dev. 19, 3043–3054 (2005).
71. Inomata, K. etal. Genotoxic stress abrogates renewal of melanocyte stem cells by triggeringtheir differentiation. Cell 137,1088–1099 (2009).
72. Mostoslavsky,R. et al. Genomic instability and aging-like phenotype in the absence ofmammalian SIRT6. Cell 124,315–329 (2006).
73. Michishita, E.et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomericchromatin. Nature 452, 492–496(2008).
74. Vousden, K. H.& Lane, D. P. p53 in health and disease. Nature Rev. Mol. Cell Biol.8, 275–283 (2007).
75. Flores, I. &Blasco, M. A. A p53-dependent response limits epidermal stem cell functionalityand organismal size in mice with short telomeres. PLoS ONE 4, e4934 (2009).
76. Artandi, S. E.et al. Telomere dysfunction promotes non-reciprocal translocations and epithelialcancers in mice. Nature 406,641–645 (2000).
77. Khoo, C. M.,Carrasco, D. R., Bosenberg, M. W., Paik, J. H. & Depinho, R. A. Ink4a/Arf tumorsuppressor does not modulate the degenerative conditions or tumor spectrum of thetelomerase-deficient mouse. Proc. Natl Acad. Sci. USA 104, 3931–3936 (2007).
78. Chambers, S.M. et al. Aging hematopoietic stem cells decline in function and exhibitepigenetic dysregulation. PLoS Biol. 5, e201 (2007).
79. Donehower, L.A. & Lozano, G. 20 years studying p53 functions in genetically engineered mice.Nature Rev. Cancer 9, 831–841(2009).
80. Matheu, A., Maraver,A. & Serrano, M. The Arf/p53 pathway in cancer and aging. Cancer Res.68, 6031–6034 (2008).
81. Matheu, A. etal. Delayed ageing through damage protection by the Arf/p53 pathway. Nature448, 375–379 (2007).
82. Mendrysa, S.M. et al. Tumor suppression and normal aging in mice with constitutivelyhigh p53 activity. Genes Dev. 20,16–21 (2006).
83. Serrano, M. &Blasco, M. A. Cancer and ageing: convergent and divergent mechanisms. NatureRev. Mol. Cell Biol. 8, 715–722(2007).
84. Begus-Nahrmann,Y. et al. p53 deletion impairs clearance of chromosomal-instable stem cellsin aging telomere-dysfunctional mice. Nature Genet. 41, 1138–1143 (2009).
85. Leri, A. etal. Ablation of telomerase and telomere loss leads to cardiac dilatation andheart failure associated with p53 upregulation. EMBO J. 22, 131–139 (2003).
86. Sablina, A. A.et al. The antioxidant function of the p53 tumor suppressor. Nature Med.11, 1306–1313 (2005).
87. Finkel, T., Deng,C. X. & Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins.Nature 460, 587–591 (2009).
88. Trifunovic, A.et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase.Nature 429, 417–423 (2004).
89. St-Pierre, J.et al. Suppression of reactive oxygen species and neurodegeneration by thePGC-1 transcriptional coactivators. Cell 127, 397–408 (2006).
本文作者认为PGC-1α为ROS防御系统强大的正调节因子,而PGC-1α缺陷可致ROS诱导性神经变性。
90. Vianna, C. R.et al. Hypomorphic mutation of PGC-1β causes mitochondrial dysfunction andliver insulin resistance. Cell Metab. 4, 453–464 (2006).
91. Vijg, J. Agingof the Genome: The Dual Role of DNA in Life and Death (Oxford Univ. Press, 2007).
92. Ruzankina, Y.et al. Deletion of the developmentally essential gene ATR in adultmice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 (2007).
93. Ito, K. etal. Regulation of oxidative stress by ATM is required for self-renewal of haematopoieticstem cells. Nature 431, 997–1002(2004).
94. Tomas-Loba, A.et al. Telomerase reverse transcriptase delays aging in cancer-resistantmice. Cell 135, 609–622 (2008).
95. Coviello-McLaughlin,G. M. & Prowse, K. R. Telomere length regulation during postnatal developmentand ageing in Mus spretus. Nucleic Acids Res. 25, 3051–3058 (1997).
96. Flores, I. etal. The longest telomeres: a general signature of adult stem cell compartments.Genes Dev. 22, 654–667 (2008).
97. Armanios, M.et al. Short telomeres are sufficient to cause the degenerative defects associatedwith aging. Am. J. Hum. Genet. 85,823–832 (2009).
98. Hao, L. Y. etal. Short telomeres, even in the presence of telomerase, limit tissue renewalcapacity. Cell 123, 1121–1131(2005).
99. Martin, G. M.Genetic modulation of senescent phenotypes in Homo sapiens. Cell 120, 523–532 (2005).
志谢
感谢L. Chin、N. Sharpless、S. Artandi、F. Muller、V. Walsh、S. Colla、M. Ugolotti、M. Jaskelioff、D. Liu 和A.-J. Chen参与讨论和评阅原稿。A. Protopopov 和E. Ivanova热心提供了图2的染色体和端粒分析。因篇幅所限,部分学界同仁的研究未能注明出处,谨致歉意。本项工作资金支持单位(编号):美国国立卫生研究院(NIH)(RO1CA84628 和U01 CA84313)、美国国防部(W81XWH-08-1-0133)、埃里森(Ellison)医学基金会。R.A.D为美国癌症学会研究教授,受Robert A. 和Renée E. Belfer基金会创新性癌症科学研究所资助。E.S.受德国科学基金会资助。
作者信息
复印和许可信息参见http://www.nature.com/reprints。作者声明不存在经济利益冲突。通信请与作者联系(Email:ron_depinho@dfci.harvard.edu)
衰老过程中端粒、线粒体及干细胞功能衰退的相关性(Linking functional decline of te.pdf
https://blog.sciencenet.cn/blog-426290-905031.html
上一篇:脑老化机制与认知衰退Neural ageing and cognitive decline
下一篇:β-淀粉样蛋白和tau蛋白在阿尔茨海默病中的毒性交互作用
全部作者的精选博文
全部作者的其他最新博文
全部精选博文导读
相关博文
- • Minerals线下恳谈会:履践致远、与时偕行——对话中国科学院广州地球化学研究所期刊合作学者
- • 聚英才 建高地 | 北京理工大学“特立青年学者”全球招聘开启
- • 700年后日本或濒临灭绝?日本学者推算预测:届时或仅剩1名15岁以下孩子
- • [转载]【同位素视角】非英语母语学者如何区分’e.g.’, ‘i.e.’, ‘namely’与‘such as’等混淆难题
- • 美国佐治亚大学等机构学者:刈割策略对Bulldog 805紫花苜蓿+Tifton 85狗牙根混播草地产量及品质的影响
- • 美国堪萨斯州立大学、密苏里大学等机构学者研究成果:土壤水分管理策略和品种多样性对紫花苜蓿产量、营养品质和农场盈利能力的影