博文
利用下代测序检测样品中的病原体
|
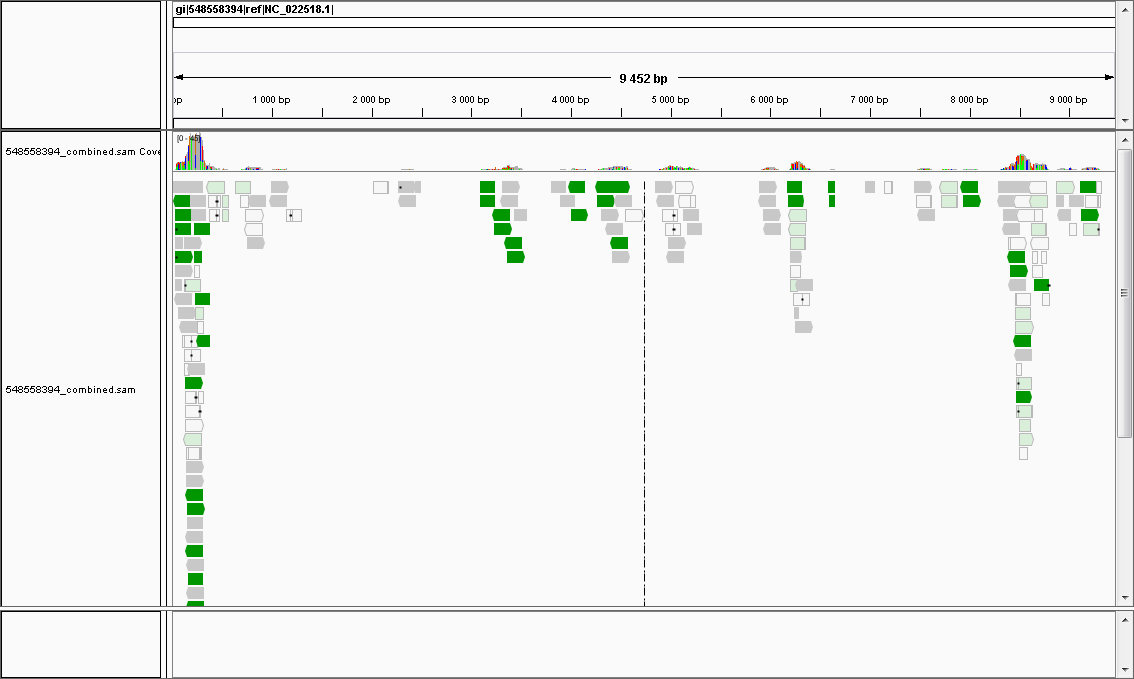
最近我的工作台上总是出现一些奇形怪状的东西。上个月,组里从隔壁医院得到了一块肝组织,是一位小朋友换肝手术之后留下的。这块肝很奇怪,因为直到最后换肝的时候,大家都不知道这位小朋友的肝到底出了什么问题。大家非常疑惑,于是就把这块肝取了样品测了序,扔给我一堆测序结果,想让我找找其中有没有可疑的病原体。前两天又有一个疑似脑炎的病人,医生在她的脑脊液中发现了和一种病毒的抗原类似的蛋白,可是却没在里面检测到这种病毒的核酸结构,这让大家都很头疼(幸好不是脑炎的那种头疼啦)。现在一堆脑脊液的测序数据又堆在了我面前,搞得我也有点头疼了。 病人到了医生那里,医生首先会想办法弄清楚病因,好对症下药。那么所有的病都是病原体引起的吗,那倒不是。像风湿,糖尿病和大多数癌症之类的病就是自身引发的,没有病原体。可是由病原体入侵人体的免疫系统导致的疾病也不在少数,平常的头疼发烧,咳嗽腹泻之类的症状大都是由病原体引起的。和大多数自身免疫病比起来,由病原体引起的病治疗起来一般会容易一点,只要鉴定出病原体的种类,然后想办法把他们从身体里除掉就好了。传统临床上鉴定样品里病原体的方法有很多。可以纯化培养,或者用病原体的DNA进行特异性扩增然后电泳或者测序,或者直接检测病人血清里的抗体类型等等。这些方法简单实用,并且能对付绝大多数的日常案例,比如说有商业化的试剂盒可以在几小时之内从呼吸道黏膜的样品里检测所有能感染呼吸道的病原体。这是因为感染特定的组织病原体往往也就那几种,医生早已对它们了如指掌,所以通常医生做的是一道选择题。但是传统的方法都没能达到很高的精确度,很难解决更复杂一点的实际问题。比如说同时确定多种病原体的存在,在样品中发现可疑的未知病原体,或者将目标病原体的基因组精确到单个碱基等等。在这种情况下,检测病原体的过程就更像是在做一道证明题了。 快速和精确地检测临床样品中的病原体不仅可以指导单个患者的治疗进程,而且也能随时监测病原体的进化,最近几年有很多二代测序应用在防治病原体上的例子。2011年,德国爆发过一场溶血性尿毒综合征,这场导致了四千多人感染的疫情的罪魁祸首是一种变异了的大肠杆菌。在一开始,PCR和MLST分型都确定这次爆发和已知的一种大肠杆菌完全一致,并没有发现和以前的一种致病大肠杆菌到底有什么差别,直到几天以后,基因组测序发现这种大肠杆菌是已知的两种大肠杆菌基因组同源重组的产物,它的直系祖先正是十年前在德国本土发现的一个菌株,只是重组后的新型在基因组和质粒上多了几个抗性基因而已。在爆发一开始,德国怀疑从西班牙进口的小黄瓜是病原体的携带者,最后西班牙的嫌疑虽然消除了,但在短短一周的时间里可怜的西班牙小黄瓜农民们就损失掉了两亿多美元。 利用二代测序还能快速确定病原体的所有毒性和抗药性。2012年新英格兰医学杂志里在对MRSA的一个研究中就做了这样的尝试。因为MRSA是从医院里长出的细菌,是以抗药而著名的,所以每次MRSA爆发都会携带不同的抗性基因而对不同的抗生素产生免疫。为了最快地确定MRSA的毒性和抗性,他们把已知的所有的毒性基因和抗药基因都收集起来分别做成了一个毒性基因库和抗药基因库。每当有新的MRSA爆发,只要对细菌进行测序,然后和两个库的基因做对比,就能很快地对变异病原体的抗药情况和毒性情况作出归纳,对症下药自然是水到渠成。 用二代测序来对样品中的病原体进行鉴定实际上就是把测序的序列在基因组数据库里寻找匹配的过程。第一步是对样品的测序,这是在实验室里完成的。测序的样品可以是多种多样:酸奶里,鸡蛋里,皮肤表面,各种组织和分泌物等等。在将它们提DNA或者RNA测序之后,就可以开始生物信息学分析了。第二步就是在数据库里寻找匹配。首先,如果在有宿主序列干扰的情况下需要去除宿主序列,这步的详细介绍可以在这里找到。而拿到没有干扰的序列之后,就是在病毒和细菌的基因组数据库里寻找匹配了。匹配之后统计出哪些微生物的基因组被匹配到的次数最多,就能基本上确定某种微生物是不是在测序的样品里了。在直接寻找匹配之前也可以先对测序序列进行拼装,再用拼装后更长的contigs在数据库里进行检索。这里的拼装对测序长度没有严格的要求,如果数据量够大的话或许也可以拼装出病原体的基因组了。 到底是什么让一开始的那个小朋友的肝脏出问题了呢?拿到了小朋友奇怪肝脏的测序结果之后,我先把测序序列用Tophat在人的基因组上找到了所有的匹配,结果发现至少有一半的序列都是来源于人的。去掉这些序列后,我们用Velvet试着做了拼装,发现只能拼接出phi174噬菌体的全基因组。这是意料之中的,因为phi174噬菌体被我们放在要测序的RNA里用作对照,可是phi174噬菌体和小朋友的肝病没有半毛钱关系。为了在基因组数据库里寻找匹配,我们下载了NCBI里所有的病毒和细菌的全基因组,然后把这些小的基因组拼成一个大的基因组文件,用bowtie把这些序列直接map在这个大基因组里。这样map之后果然发现了样品里有几种很有趣的病毒。虽然coverage都不是很高,但是是确定存在的。就像下面这种,这是用IGV看到的其中一个病毒基因组的map结果: 虽然这个病毒基因组的有些区域没有被测序序列覆盖到,但这么多读段能map到很多不同的地方就能足以证明这种病毒是在样品里确定存在的。有些区域没有被覆盖到只是因为测序深度不够,如果再有多几倍的数据量的话拼接出这个病毒的基因组是完全有可能的。当然如果只是要鉴定病原体的存在,那么像上面这样,几乘的数据量就够了。但如果你还希望拼接出完整的基因组,根据所选的测序仪读长和准确度的不同,数据量要达到30乘甚至更高才行 。 有时候mapping结果里会有很多复杂度很小的读段被map在某个基因组上,比如说连着一百多个T,但只要简单地在SAM文件里检查一下map在这个基因组上的读段就可以发现它们其实是假阳性。同时也应当检查这些读段是不是map在了整个基因组的所有区域。如果map在一个基因组上的所有读段都只是map在这个基因组的一个或两个很小区域的话也是有问题的,就像下面这张图一样。虽然有很多reads map在了这个基因组上,但全部的reads都集中在了一个位置,而且错配不少,这最有可能是因为一个其他微生物的一个基因片断恰好与它相似,并不能说明这种微生物存在在样品里。 对样品测序选择什么样的平台才最合适呢?这要考虑到很多实际的情况。比如说测序所需要的时间,鉴定病原体所要求的精确度,以及测序的花费等等。小量的微生物测序通常会用两种测序平台,一个Miseq,另一个是ion PGM。两个机器虽然价钱差不多,但是性能有不小的差别。通常情况下可以根据精度需要来决定通量,然后根据时间成本和所需要的通量来选择合适的测序平台。有些平台比如说ion PGM系列有量小速度快的优点,而Miseq数据量适中,速度稍慢,但是每个base测得更准确,平均到每个base上也可以更省钱。当然ion系列的通量从几百M到几十G不等,这就要看研究需要了。我们实验室在仔细考虑之后还是决定买了Miseq,所以我常常调侃说那台Miseq才是真正雇我做事的人 :D 在基因组数据库里寻找匹配很多软件都可以。最主流的有Bowtie系列,BLAST或者HMMER。这三个软件各自利用完全不同的算法,但目的都是在数据库里寻找匹配。作为一个启发式算法,BLAST虽然比动态规划有着时间上的优势,但这却是以牺牲了一些精确度作为代价的。尤其是在数据库非常大的时候,它不会百分之百保证在数据库中找出与查询序列相似度最高的那段序列,所以我觉得BLAST更适合长度1000bp以上的query。HMMER用的是profile隐马可夫模型,在官网号称比BLAST要快而且精确度高,但它没有像BLAST那样有一个公共的网页用户界面,所以用的人比较少。而相比于BLAST和HMMER,bowtie是可以保证为每个读段找出最佳匹配的。用bowtie的另一个好处是可以将mapping之后的SAM文件在IGV里可视化,这样就可以很直观地检查reads在一个基因组上的覆盖情况。所以我一般是用Bowtie2来进行短reads的直接mapping,用本地BLAST对组装以后的序列进行匹配查找,这样基本上就能保证分析的准确度了。这个python程序可以自动对乱成一团的Blast,HMMER或者bowtie的mapping结果进行总结归纳,让比对结果一目了然。 可惜的是,强大的二代测序还没有在现代医疗的诊断中施展开拳脚。在非洲大多数医院连一台PCR仪都没有的情况下,买测序仪就显得太奢侈了。因此测序仪也只在一些发达地区的医院里才有配备。在对付一些疑难杂症和未知的病原体的疾病上,它的高通量虽然可以为医生提供更精准的目标病原体的信息,但真正需要如此高精确度的鉴定场合却少之又少,所以测序仍旧是有钱人才能用得起的服务,将它作为医院检测的常规手段目前还不大现实。和其他基于抗体和pcr的检测手段比起来,测序不仅需要长时间的前期工作,而且需要更复杂的后期分析,一个样品从头到尾成本少说也要几百美元,快马加鞭也要一天才能出结果。而一个基于抗体检测的商业试剂盒在几个小时内就可以完成特定样品里常见病原体的检测,每个样品的成本却只有几美元,大批量处理起来非常占优势。二代测序虽然输在了成本上,可是它的灵敏度和终极的精确度也会让它成为临床检测的一个重要的方法。 和医院日常检测相比,二代测序的更大功用还是发挥在了科研领域。比如说疑难杂症的治疗,研究病原体的侵染习性,对未知微生物世界的探索等等。而二代测序可以在科学家在对样品知之甚少的情况下提供很多线索,更有可能拿到新的基因组。这不仅有科学价值,而且在抗击动植物病原体上也有很大的潜在经济价值。虽然成本不低,但如果用对了地方,二代测序也是能很有一番作为的。

https://blog.sciencenet.cn/blog-635619-921701.html
上一篇:用哈希高效实现NGS序列的k-mer检测
全部作者的精选博文
- • RNA病毒基因组组装指南
- • 唾液比你更懂你?
- • 亚马逊雨林里的向导们
- • 热带雨林离我们有多远
- • 你的实验室该买哪种测序仪?
- • C值悖论是由进化造成的吗?