博文
宏基因组/转录组去除宿主污染
||
对于宿主(动植物)来源的宏基因组、(宏)转录组,去除宿主序列污染对于序列拼接等分析的准确定至关重要,我们可以通过将raw reads或初步质控后的clean reads与宿主的基因组序列进行多重比对,提取其中未比对上的序列,就可以获得去除了宿主序列的reads。
由于做人体微生物的比较多,所以这里以人宿主污染为例。
1、下载人类基因组,构建bowtie比对数据库
在https://hgdownload.soe.ucsc.edu/goldenPath/hg19/chromosomes/可以找到hg19的人基因组序列数据。
wget --timestamping 'ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/*'
下载所有序列数据,或者只下载23对染色体的序列数据也就是ch'r*.fa.gz.
tar -xzvf * | cat > hg19.fa
解压所有序列并合并为一个fasta文件。
使用bowtie2的bowtie-build命令构建比对数据库:
bowtie2-build hg19.fa hg19
2、使用bowtie进行多重比对,当然这里也可以选择bwa之类的工具也都可以。
bowtie2 -x hg19 -p 90 -1 lib1 -2 lib2 -S host_mapped.sam
-x为比对数据库,如果不在当前路径前面要加上路径信息。
-p为使用多少个线程,我这里用的90
-1 -2后面是双端序列的两个fastq文件
-S为比对结果的sam文件
3、用samtoos对比对结果进行格式转换,提取其中的未比对序列。
首先将二进制的sam文件转为bam格式的文本文件
samtools view -@ 10 -bS host_mapped.sam > host_mapped.bam
-@为使用的线程数
-b表示输出bam格式 S表示自动检测输入文件格式
结果通过>重定向保存成一个bam文件
然后将未比对上的序列提取出来
samtools view -b -@ 10 -f 12 -F 256 host_mapped.bam > host_unmapped.bam
-f 和-F通过比对结果的flag信息共同将比对或未比对上的序列提取出来, 这一部分可以看一下samtools的说明。-f 表示只包含有后面该flag信息的序列。因为我们是双端序列,这里我们应该输入的数值是4+8,也就是12.-F表示仅包含没有该flag的序列,这里我们不需要比对上的序列,也就是256.标签的含义见下表。
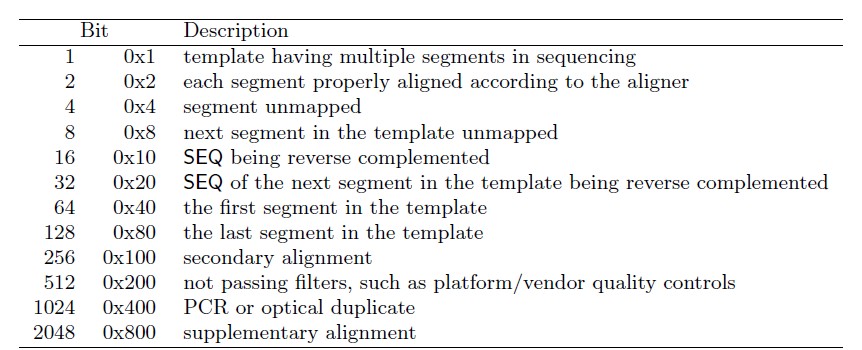
最后将bam文件排下序整理一下。
samtools sort -@ 10 -n host_unmapped.bam host_unmapped_sorted.bam
4、用bedtools将bam文件转为fastq格式的序列数据。
最后一步是将提取并排序好的bam文件中的序列和质量信息提取出来,转换成fastq格式的数据,使用bedtools工具来完成。
bedtools bamtofastq -i host_unmapped_sorted.bam -fq host_removed_1.fq -fq2 host_removed_2.fq
-fq和-fq2分别表示正反向序列。
到这里就完成了序列的宿主过滤。其他宿主除了参考基因组不一样,其余过程都是相同的。
https://blog.sciencenet.cn/blog-2379401-1268509.html
上一篇:使用Aspera批量下载GeneBank和EMBL数据库数据