博文
CASTEP文献解读丨厦大黄小青Nat Commun:亚纳米高熵合金纳米线实现卓越的氢氧化催化
||

具有独特理化性质的高熵合金(HEAs)在许多领域引起了极大的关注,但在原子水平上对尺寸和形貌的精确控制仍然是艰巨的挑战。在此,厦门大学黄小青教授和卜令正教授、香港理工大学黄勃龙教授(共同通讯作者)等人合成了用于碱性氢氧化反应(HOR)的独特PtRuNiCoFeMo HEA亚纳米纳米线(SNW)。HEA SNWs/C的质量和比活分别达到6.75 A mgPt+Ru-1和8.96 mA cm-2,分别是HEA NPs/C、商用PtRu/C和Pt/C的2.8/2.6、4.1/2.4和19.8/18.7倍。此外,在加速耐久性试验 (ADT) 中,2000 次循环后没有观察到 HOR 性能的明显衰减,表明 HEA SNW 对 HOR 的稳定性非常好。在1000 ppm CO存在的情况下,它甚至可以在HOR期间显示出增强的抗CO中毒能力。密度泛函理论计算表明,HEA SNWs中不同金属位点之间的强相互作用可以极大地调节质子和羟基的结合强度,从而增强HOR活性。这项工作不仅为制备Pt基HEA亚纳米/纳米材料提供了可行的合成途径,而且促进了催化及其他方面的基础研究。

图1.HEA SNWs的表征。(a) HAADF-STEM图像。(b) XRD图。(C)晶体结构。(d) SEM-EDS和不同元素的原子比。(e) HAADF-STEM-EDS元素映射。(f-h)像差校正的HRSTEM图像。(i)3D模型和HEA SNW的放大原子模型。
他们采用了乙酰丙酮铂(II),乙酰丙酮钌(III),乙酰丙酮镍(II),钴(III)乙酰丙酮化物、乙酰丙酮铁(III)和六羰基钼作为金属前体,油胺作为溶剂,硬脂基三甲基铵溴化物作为结构导向剂,葡萄糖作为还原剂合成了HEA SNW。透射电子显微镜和高角度环形暗场扫描图像显示获得的HEA SNW 的平均直径为1.8 ± 0.3 nm。X射线衍射(XRD)图中的特征峰归因于Pt的面心立方(fcc)结构(JCPDS No.04-0802),峰的正移意味着合金的形成。
另一方面,XRD图中峰的减弱和加宽可归因于HEA SNW中的晶格畸变。扫描电子显微镜能量色散X射线光谱(SEM-EDS)表明HEA SNWs的组成为Pt/Ru/Ni/Co/Fe/Mo = 29.6/9.3/15.2/15.6/12.2/18.1,这与电感耦合等离子体发射光谱(ICP-OES)测量结果相近(Pt/Ru/Ni/Co/Fe/Mo = 28.5/6.8/18.6/15.2/10.7/20.2。此外,HAADF-STEM-EDS元素映射和线扫描分析表明,所有金属均匀分布在HEA SNW中。此外,像差校正的高分辨率STEM(HRSTEM)图像显示获得的HEA SNW具有扭曲的结构,具有丰富的原子台阶和富含缺陷的晶格失配。此外, 3D原子模型来描绘HEA SNW的结构,生动地揭示了具有丰富表面原子台阶和众多小平面边界的HEA SNW的独特结构。
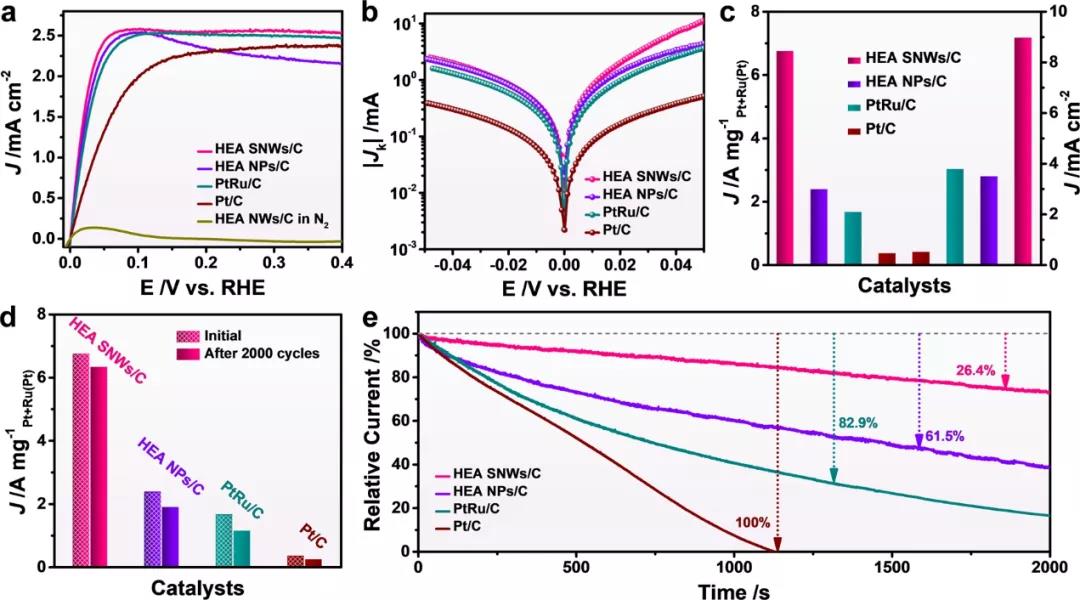
图 2:HEA SNWs/C和其他催化剂的HOR性能评估。(a)在H2饱和的0.1 M KOH中的极化曲线。(b)塔菲尔图。(c)相对于RHE,在50 mV过电位下的标准化质量活性和比活性。(d) 2000次ADT 循环前后的标准化质量活性和比活性。(e)不同催化剂在 1000 ppm CO/H2 饱和的0.1 M KOH中在100 mV与RHE下的相对电流-时间计时电流响应。
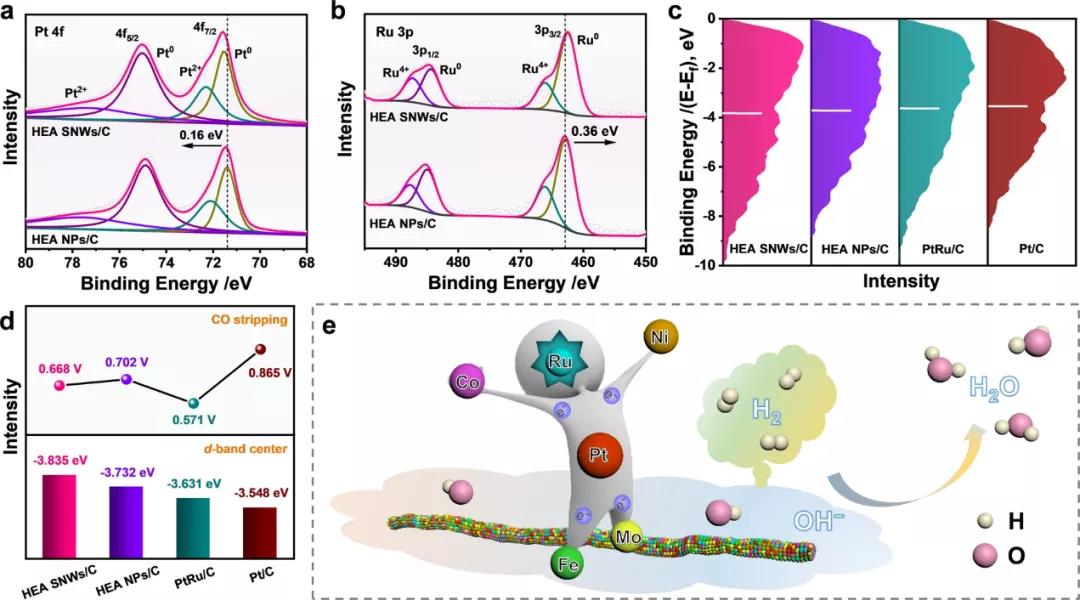
图 3:表面价带光电发射光谱分析。(a)Pt 4f XPS光谱。(b) Ru 3p XPS光谱。(c)不同催化剂的d带中心。垂直线表示样品相对于费米能级的d带中心。(d)不同催化剂的CO剥离电位和d带中心位置。(e) HOR在HEA SNWs/C的示意图。
为了揭示在HEA SNWs/C上增强HOR性能的机制,通过X射线光电子能谱(XPS)测量研究了催化剂的表面性质。值得注意的是,HEA SNWs中的所有金属元素都以金属态和氧化态混合。与HEA NPs/C相比,HEA SNWs/C的Pt0 4 f XPS光谱中的峰正移0.13 eV,而Ru0 3p XPS光谱中的峰负移0.36 eV,表明在 HEA SNW中电子可能从Pt转移到Ru。
另一方面,与HEA NP相比,HEA SNW的Ni0 2p、Co0 2p、Fe0 2p 和 Mo4+ 3d XPS光谱分别正向偏移 0.26、0.17、0.93 和 0.25 eV。考虑到只有Ru0 3p XPS光谱负移0.36 eV,可以得出结论,Ni0 2p、Co0 2p、Fe0 2p 和 Mo4+ 3d XPS光谱的正移归因于电子从Ni、Co、Fe和Mo转移到HEA SNW中的Ru。
为了进一步研究电子转移对HOR过程中反应物和中间体结合强度的影响,进一步比较了各种催化剂的d带中心。与HEA NPs/C (-3.732 eV)、PtRu/C (-3.631 eV)和Pt/C (-3.548 eV)的值相比,HEA SNWs/C的d波段中心向上移动至-3.835 eV,从而削弱了H的吸附,但促进了OH中间体的吸附28,29。考虑到Ru对HOR的显着影响,可以促进OH*物质的吸附,我们评估了HEA SNWs和PtNiCoFeMo HEA SNWs/C的HOR性能。HEA SNWs/C的质量活性是PtNiCoFeMo HEA SNWs/C的11.8倍。
此外,与PtNiCoFeMo HEA SNWs/C相比,HEA SNWs/C对CO中毒的抵抗力显着提高。特别是,当使用HEA SNWs/C作为催化剂时,在1000 ppm CO存在下的HOR活性在2000 s后下降了26.4%,这远小于PtNiCoFeMo HEA SNWs/C (85.2%),证实了显着性河上的Ru。此外,与Pt/C相比,HEA SNWs/C的CO剥离峰的位置正向移动,这进一步表明Ru的存在显着提高了对CO中毒的抵抗力。上述结果意味着HEA SNW中不同元素之间的强电子相互作用可以调节H和OH物种的吸附能力,进而增强碱性HOR活性。
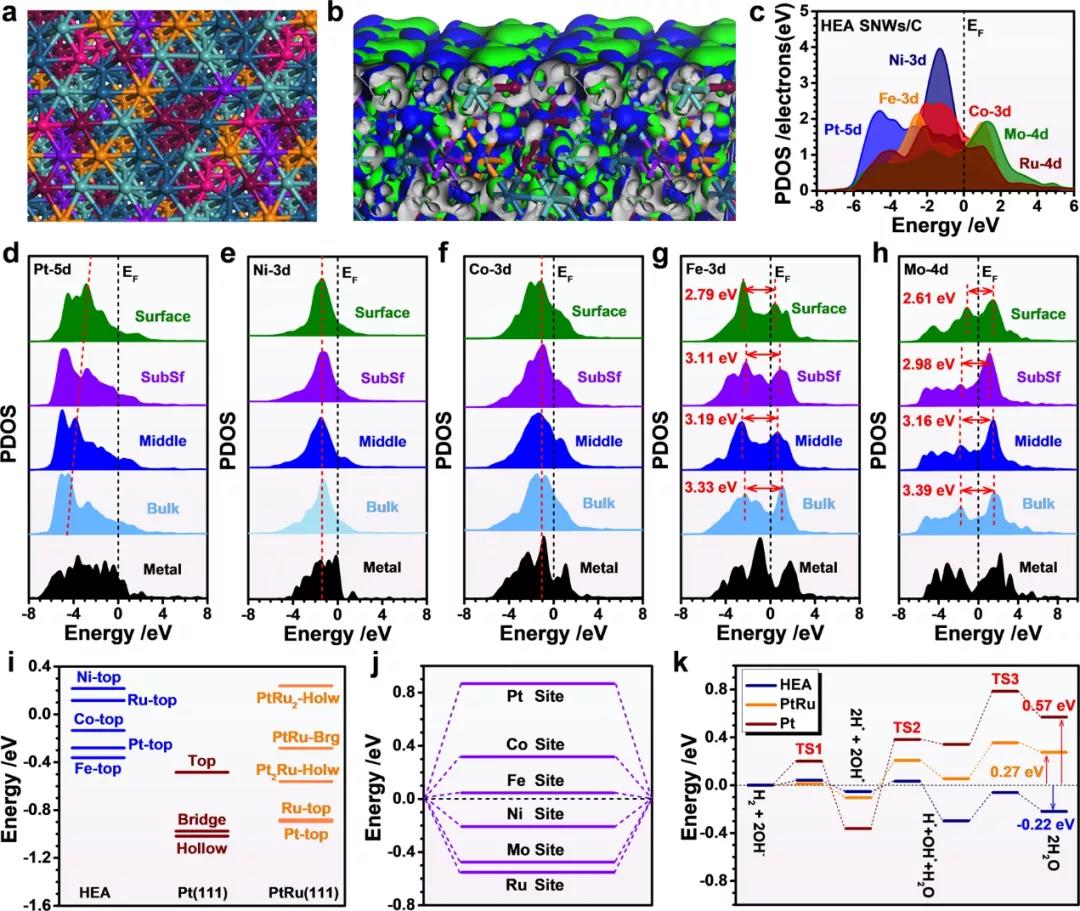
图 4:HEA SNW上HOR路径的DFT计算。(a)几何优化后的俯视图。深绿色、棕色、紫色、橙色、浅绿色和粉红色的球分别代表 Pt、Ni、Co、Fe、Mo 和 Ru 原子。(b)费米能级附近电子分布的3D等高线图。(c)PDOS。(d)Pt-5d、(e)Ni-3d、(f)Co-3d、(g)Fe-3d 和(h)Mo-4d的位点依赖PDOS。(i)HEA SNWs/C、Pt (111)和PtRu (111)之间的HBE比较。(j)HEA SNWs/C上的OH结合能。(k)HOR在 HEA SNWs/C、Pt (111)和PtRu (111) 上的能量趋势。
采用CASTEP进行DFT计算以进一步研究增强HOR性能的机制。值得注意的是,Ni和Ru位点都显示出细微的扭曲,而整体HEA SNWs保持稳定,这证实了HEA SNWs/C的稳定性。在电子结构方面,HEA表面上键合轨道和反键合轨道之间的强耦合提供了高HOR活性,并在不同金属位点之间实现了有效的位点到位点电子转移。
此外,HEA SNW的详细电子结构也通过投影局部状态密度(PDOS)来证明。观察到d轨道之间明显的重叠,表明不同金属之间的强键合。特别是,明显尖锐的Ni 3d轨道位于-1.30 eV附近,这是H*和OH* 强吸附的关键指标。同时,Co 3d轨道显示出与Ni 3d轨道相似的位置,这可能进一步促进电子转移。对于Fe 3d和Mo 4d轨道也观察到了类似的现象,d轨道的重叠不仅可以增强电子转移,而且还可以对Ni和Co位点的3d轨道施加钉扎效应,以获得强大的电活性。
另一方面,Pt 5d轨道位于费米能级最远的位置,表明Ru在电催化过程中充当电子库以平衡HEA NW的价态。此外,Ru 4d的范围从-6.0到 +4.0 eV,这可以实现灵活的电子调制并加速HEA 表面上的点到点电子转移。此外,已经证明了HEA SNW中每个元素的位点相关电子结构。如图4d所示,Pt-5d中心从主体(青色)到 HEA SNWs的表面(绿色)逐渐接近费米能级,表明H2吸附在表面上比在表面上强得多。大部分HEA SNW。与纯Pt金属(黑色)相比,整体Pt-5d轨道远离费米能级,表明HEA SNWs具有适当水平的Pt-5d带用于H2吸附,因此表现出增强的HOR活性.纯Ni金属(黑色)的Ni-3d轨道比HEA SNWs的Ni-3d轨道宽得多,表明HEA SNWs中Ni-3d轨道的局域电子密度远高于纯Ni金属,这可以为电荷转移和与OH*的键合机会带来更多的可能性。
此外,从本体到表面,Co 3d轨道的eg-t2g带分裂逐渐消失导致轨道内eg-t2g电子转移有效,而不是在dd轨道之间转移,这进一步促进了位点到-从催化剂到吸附物的位点电子转移。还注意到,HEA SNW中Co的电子结构从本体到表面都相似。这种强大的电子结构归因于来自相邻Fe/Mo 和Ru轨道的屏蔽效应的双重钉扎效应。对于Fe位点,3d 轨道的eg-t2g带分裂从体位点到表面位点从3.33减少到2.79 eV,支持更有效的电子转移到OH*。对于Mo-4d轨道,观察到eg-t2g带分裂的类似降低,并且能量势垒从本体到表面从3.39显着降低到2.61 eV。HEA SNWs放宽了轨道内eg-t2g电子转移的禁律,消除了Fe-3d能带和Mo-4d能带的eg-t2g分裂间隙。带之间较小的间隙进一步降低了表面上位点到位点电子转移的势垒。Fe和Mo位点中电子结构的演变显示出类似的趋势,即eg-t2g分裂的减少。这不仅改善了表面的位点电子转移,而且加强了价态对Ni和Co位点的保护作用。此外,我们对电子结构进行了更详细的表征,并证实了HEA表面电活性的提高,这保证了有效的HOR。
此外,还比较了HEA SNWs/C的氢结合能(HBE)和羟基结合能(OHBE)。与H*在Pt (111)和PtRu (111)上的强结合相比,H*在HEA SNWs/C上的结合强度已显着减弱到接近0 eV(H* 结合的理想值)的适当水平,导致HOR活性高于HEA SNWs/C。对于OHBE,尽管OH*结合强度显示出Pt < Co < Fe < Ni < Mo < Ru的趋势,但OH*在Ru上的结合能接近-0.6 eV,表明OH*在HEA SNWs上的吸附适度/C。众所周知,吸附的OH*在去除以H2O形式吸附的质子方面起着至关重要的作用,因此显着增强了碱性HOR活性。另一方面,HEA上的整体反应是放热的,释放的能量为0.22 eV,表明H2O的快速解吸。与此形成鲜明对比的是,Pt 和PtRu上的整体反应是吸热的,能量分别为0.57和0.27 eV。请注意,HEA表面形成H2O的能垒低至0.08 eV,远低于Pt和PtRu,进一步证实了HEA的HOR活性优于Pt和PtRu。
Changhong Zhan, Yong Xu, Lingzheng Bu, Huaze Zhu, Yonggang Feng, Tang Yang, Ying Zhang, Zhiqing Yang, Bolong Huang, Qi Shao & Xiaoqing Huang. Subnanometer high-entropy alloy nanowires enable remarkable hydrogen oxidation catalysis. Nature Communications
文章链接:
https://www.nature.com/articles/s41467-021-26425-2#Sec13
Materials Studio是久负盛名计算模拟软件,问世20余年来,经过不断地迭代优化,使其功能异常强大,极易上手,初学者只需通过简单的参数设置和点击鼠标就能完成DFT计算。其计算可靠性久经考验,备受Nature、Science等顶级期刊认可。
华算科技和Materials Studio官方代理深圳浦华系统联合推出Materials Studio建模、计算、分析课程。课程专为零基础学员设计,沿着理论讲解、模型搭建、性质计算、结果分析层层递进讲解,带你快速入门DFT计算。课程极度注重实操,全程线上直播,提供无限回放,课程群在线答疑。(详情点击下方图片跳转)
识别下方二维码报名,或者联系手机13005427160。

https://blog.sciencenet.cn/blog-2531381-1314550.html
上一篇:CASTEP相关文献解读丨香港理工黄勃龙Small:Pr3+:异质结构 Li1−xNaxNbO3中的有效可重复机械发光
下一篇:入门宝藏!MS公开课视频、自制脚本、官方教程中文版、答疑手册下载链接
全部作者的其他最新博文
- • Materials Studio官方教程:Mesocite——磷脂双分子层的粗颗粒化分子动力学【1】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【3】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【2】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【1】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【3】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【2】