博文
Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【3】
||

Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【1】
Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【2】
7、运行模拟
在运行模拟之前,需要一个新的构型文档。在上一个教程中生成的构型文档是不够的,因为与以前的每个晶格单元仅有一个位点相比,现在每个晶格单元有三个位点。
单击Modules工具栏上的Kinetix按钮![]() ,然后从下拉列表中选择Configuration Builder。
,然后从下拉列表中选择Configuration Builder。
将Configuration size更改为64×64。确保Type of configuration设置为Constant,并从Adsorption sites下拉列表中选择V。单击Create按钮并关闭对话框。
将创建一个新文档KinetixConfiguration.xkc。注意,由于每个晶格单元具有多个位点,因此这与上一教程中的显示不同。
将新创建的构型文件重命名为ZGBMultiSite.xkc,保存工程。
单击Modules工具栏上的Kinetix![]() 按钮,然后从下拉列表中选择Calculation,或从菜单栏中选择Modules | Kinetix | Calculation。
按钮,然后从下拉列表中选择Calculation,或从菜单栏中选择Modules | Kinetix | Calculation。
在本教程中,未指定显式速率系数(吸附过程除外)。使用Arrhenius表达式,速率系数高度依赖于温度。因此,需要指定模拟所需的温度。可以使用Temperature programmed任务设置温度变化过程,但在本教程中,将使用恒温。
与前面的教程相比,此例中速率系数非常高。这意味着模拟时间Simulation time和采样间隔Sampling interval的默认值不再合适。模拟的速度要比默认值快得多,将在纳秒级的时间尺度完成。在实际操作中,可以通过以更短的模拟时间和采样间隔运行模拟,逐渐增加采样间隔,直到每个采样间隔中出现有统计意义的反应数。
确保ZGBMultiSite.xkc为当前文档,并选择了Constant conditions任务。单击More...按钮,打开Kinetix Constant Conditions对话框。
将Temperature设置为1200 K。将Simulation time设置为1.0e-7 s,Sampling interval设置为1.0e-9 s,关闭对话框。
在Setup选项卡中,从Processes下拉列表中选择ZGBMultiSite.xkp。
单击Run按钮,关闭对话框。
将在Project Explorer中打开一个新的名为ZGBMultiSite Kinetix ConstCond的文件夹。当计算任务完成后,将创建与上个教程中相同的一组文档。
8、分析结果
在Project Explorer中,从新的结果文件夹中打开ZGBMultiSite.kout文件。
单击Modules工具栏上的Kinetix按钮![]() ,然后从下拉列表中选择Analysis,打开Kinetix Analysis对话框。
,然后从下拉列表中选择Analysis,打开Kinetix Analysis对话框。
选择Concentrations,勾选CO和O Species复选框。单击View按钮。
将产生如下图所示的曲线图:
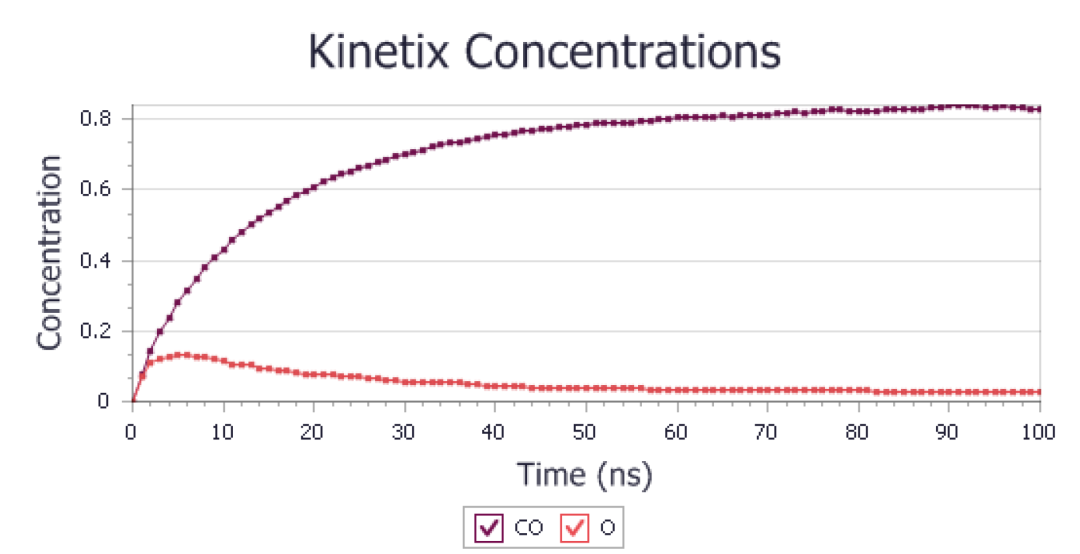
模拟过程中化合物的平均浓度
在Kinetix Analysis对话框中,选择Rates并勾选所有Reactions复选框。单击View按钮。
由此产生的图表将立即示出,模拟主要由两个扩散过程控制。现在,将生成另一个没有扩散过程的速率图。
在Kinetix Analysis对话框中,取消勾选CODiff、ODiff_fh和ODiff_hf复选框。单击View按钮。然后关闭对话框。
将产生如下图所示的曲线图:

模拟过程中的平均反应速率
现在应查看新的构型演变过程。
在Project Explorer中,从新的结果文件夹中打开ZGBMultiSiteTraj.xkc文件。使用animation controls查看每帧构型。停止查看轨迹动画。
图表文档和构型的轨迹文档可通过在图表中选择点进行链接。当逐步查看构型轨迹中的每帧时,将在每个打开的图表中的一条曲线上示出表示相应时间的点。相反,如果在其中一个图表中选择了一个点,构型轨迹将改变,以显示最接近所选点的时间的一帧。
应该能够确认禁止占用位点的正确设置。每个CO或O化合物都应该被空位完全包围。现在,可以更改显示以强调这些化合物。
在ZGBMultiSiteTraj.xkc中右键单击,从弹出的快捷菜单中选择Display Style,打开Kinetix Configuration Display Style对话框。
在Options选项卡中,将Background color更改为white。在Style选项卡中,将Radius scale更改为1.3。
关闭对话框。
新的ZGBMultiSiteTraj.xkc视图应近似如下所示:

模拟过程中的构型
此示例显示了模拟的初始化阶段。可以单击Configuration Viewer工具栏上的按钮,逐步浏览每一帧构型;在计算结束时,位点占用率代表稳态条件。本教程中用于模拟的模型比Pt(1 1 1)表面CO氧化的简单模拟(Simple modeling of CO oxidation on a Pt(1 1 1) surface)教程中使用的模型更复杂。考虑了多个位点及其在反应中的不同作用。使用Arrhenius表达式指定反应速率,给出温度依赖性。但是,还可以添加另一个复杂过程,即横向相互作用,以使模型更加真实。
9、添加横向相互作用
横向相互作用模拟了附近位点的化合物对过程(改变活化能)的影响。
在Kinetix中,横向相互作用通过指定一个过程中相关化合物及其邻近化合物组合的活化能变化来描述。当利用服务器进行计算,创建输入文件时,将合并到过程的最终描述中。
相互作用对于关联的化合物是对称的。因此,当与B相邻时涉及化合物A的过程的活化能变化,和与A相邻时涉及化合物B的类似过程相同。
fcc位点上氧原子之间的横向相互作用值已通过密度泛函理论计算得出(Jansen et al., 2005)。假设hcp位点的相互作用具有相同的值。CO分子间相互作用的数值取自kMC模拟的CO/Rh(100)程序升温脱附实验的拟合值(Jansen, 2004)。根据位点之间的距离和类似距离的其他体系的横向相互作用值,对不同穴位处CO和氧之间以及氧原子之间的相互作用值进行了估算。
对于相互作用中涉及的两种化合物之间的不同组合方式,需要添加三组横向相互作用。首先,将为CO-CO组合添加相互作用。
在原始ZGBMultiSite.xkp文件的Interactions选项卡中,从每个Interactions下拉列表中选择CO,设置cutoff为3.5 Å。单击Add按钮。
新的相互作用将添加到此选项卡的列表中,详细信息列在Interaction Details选项卡上。列出了最小和最大截止距离之间所有可能的位点组合。其中一些是与体系不相关的,例如,CO只能存在于顶位(位点索引为1)。
列出的许多相互作用详细信息通过晶格定义的对称性相互关联。虽然这些关系未显示在列表中,但在修改列表时,例如通过删除详细信息项或修改相互作用值,应进行考虑。
应从此过程的列表中删除不相关的相互作用详细信息,并为其余详细信息指定所需的相互作用值。
在Interaction Details选项卡上,选择任一Site for CO列中位点索引为2或3的一行(位点描述的第三个组分)。按下DELETE。
重复此操作以删除除顶位之间的相互作用之外的所有相互作用,因此所有剩余相互作用两个位点的索引均为1。
注意:Interactions Details选项卡不支持选择多行。
Interaction Details列表中应剩余六项。
单击其中一个Interaction单元格,将值更改为6.69。两个对称相关的相互作用行将自动更新。
重复该过程,直到所有的Interaction值均为6.69。
接下来将添加CO和O之间的横向相互作用。此处将利用下列事实:CO只能存在于顶位,而O只能存在于穴位和fcc位。
在Interactions选项卡上,为第二个相互作用化合物选择O,然后单击Add按钮。
在Interaction Details选项卡上,选择Site for O列中位点索引为1的一行,按下DELETE键。
重复此操作,直到剩余Site for O的索引为2或3。
删除Site for CO索引为2或3的所有相互作用。
将所有Interaction值更改为2.39。
此相互作用的Interaction Details列表中应剩余六项。
接下来,将添加O和O之间的横向相互作用。这更为复杂,因为在不同距离处会有两组相互作用,因此具有不同的相互作用值。
在Interactions选项卡上,两种化合物均选择O,然后单击Add按钮。
在Interaction Details选项卡上,删除所有Site for O索引为1的行。
fcc位点之间将有六个相互作用项(位点索引为2),hcp位点之间将有六个相互作用项(位点索引为3)。这些都将显示2.77 Å的距离。同样,fcc位点和hcp位点之间也会有六个相互作用项,显示距离为3.199 Å。
对于显示距离为2.77 Å的所有相互作用项,将Interaction值更改为6.21。
对于显示距离为3.199 Å的所有相互作用项,将Interaction值更改为2.39。
Interaction Details选项卡应如下所示:
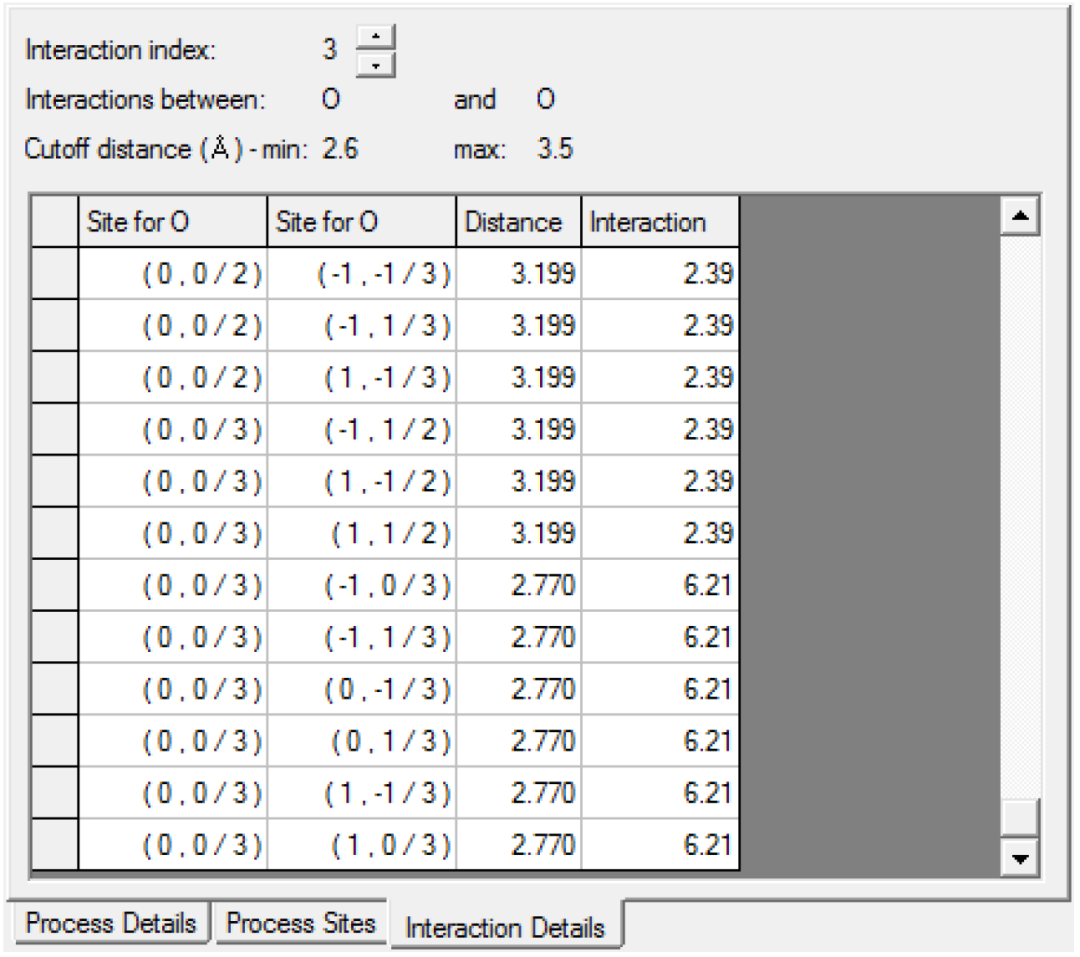
Interaction Details选项卡
在继续下一步骤之前,应保存工程。
从菜单栏中选择File | Save Project。
也可以在此步骤之后,将文档和Materials Studio示例文件夹中预先保存的文件版本(ZGBMultiSite Stage 4.xkp)进行对比。
10、应用横向相互作用并运行模拟
默认情况下,已定义的任何相互作用将用于有条件地修改使用Arrhenius表达式作为速率规定的任何过程的活化能。在本教程前面的模拟运行中,没有定义相互作用,因此没有此类修改。
对于每一个这样的过程,横向相互作用改变活化能的程度由Brønsted-Polanyi factor控制,用α表示。首次创建过程时,α被设置为一个取决于过程类型的值。该默认值对于计算目的来说是正确的,但是应该检查它们是否被意外更改。
在Process Details选项卡上,检查每个过程的详细信息。对于每个Desorption过程,应勾选Use interactions复选框,且Brønsted-Polanyi factor应为1.0。
对于每个Diffusion过程,应勾选Use interactions复选框,且Brønsted-Polanyi factor应为0.5。
现在可以运行另一个模拟计算,并将结果与未应用横向相互作用的前一个模拟进行比较。
打开Kinetix Calculation对话框。确保原始的ZGBMultiSite.xkc为当前文档,并选中了Constant conditions任务。在Job Control选项卡上取消勾选Automatic复选框,将Job description设置为ZGB MultiSite With Interactions。
单击Run按钮,关闭对话框。
11、分析结果
应创建一个Concentrations的图表,并对ZGBMultiSite With InteractionsTraj.xkc文档中的显示模式修改成与之前的模拟中所使用的显示模式相同。
应与下图相似:
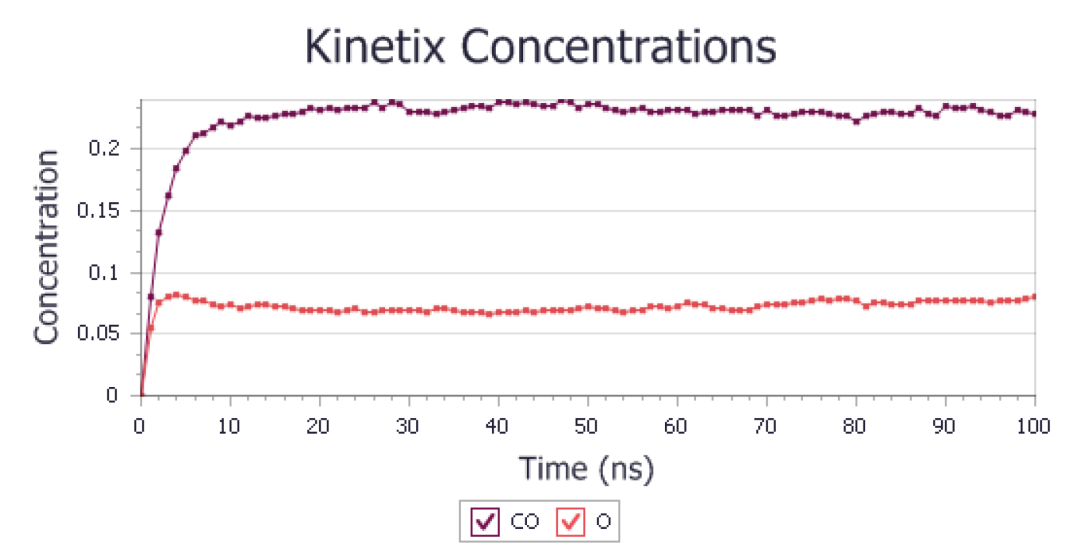
添加相互作用模拟时的平均浓度
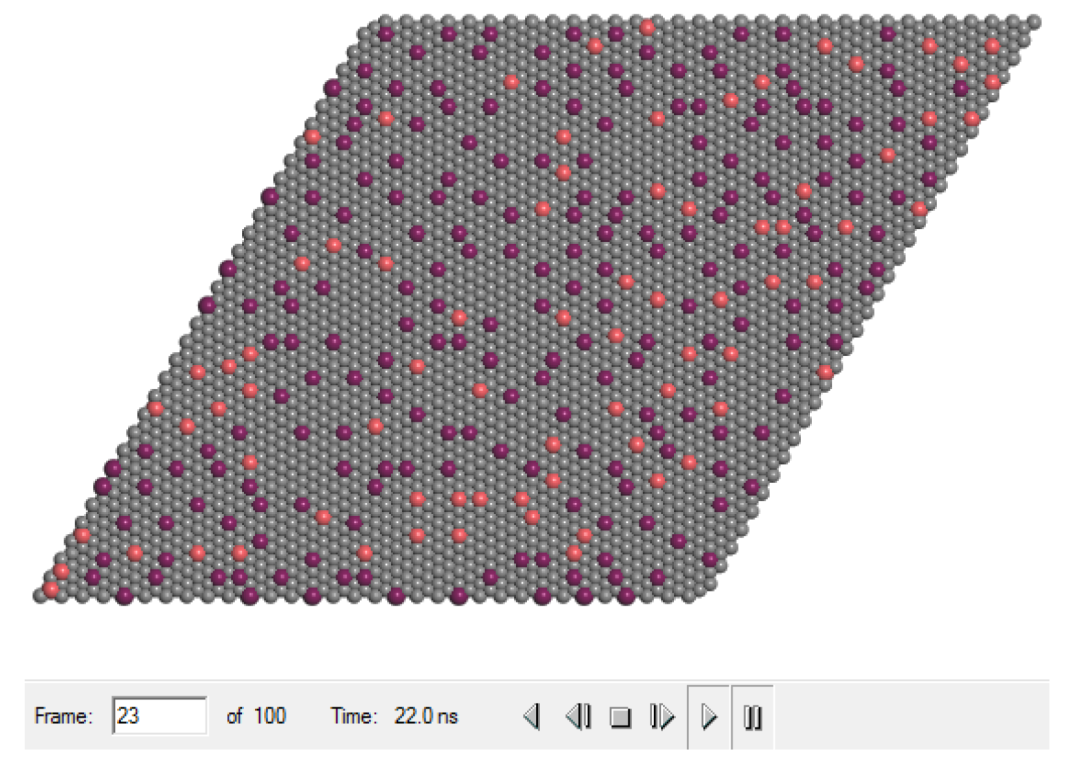
应用相互作用模拟时的构型
现在可以将这些结果与添加横向相互作用之前生成的结果进行比较。可见CO的浓度稳定在约0.23的值,远低于之前的值,并且氧浓度也稳定在约0.06,而在之前的模拟中,它下降到约0.03。
此时,可以以.avi文件的形式导出轨迹,以便在演示文稿中播放。
本教程到此结束。
本教程对应视频将在杨站长视频号、华算科技B站同步推送,敬请各位关注。
参考文献:
A.P.J. Jansen, "An introduction to Monte Carlo Simulations of Surface Reactions",
https://arxiv.org/abs/cond-mat/0303028 (2003).
A.P.J. Jansen, "Monte Carlo simulations of temperature-programmed desorption spectra",Phys. Rev. B, 69, 035414 (2004).
A.P.J. Jansen and W.K. Offermans, "Lateral interactions in O/Pt(111): Density Functional Theory and kinetic Monte Carlo", in the Proceedings of the International Conference on Computational Science and Its Applications (ICCSA-2005), (Springer, Berlin, 2005), pp. 1020-1029.
A.P.J. Jansen, "An Introduction to Kinetic Monte Carlo Simulations of Surface Reactions" (Springer, Berlin, 2012).
W.K. Offermans, A.P.J. Jansen, and R.A. van Santen, "Ammonia activation on platinum{111}; A densityfunctional theory study", Surf. Sci., 600, 1714-1734 (2006).
A.P. van Bavel, M.J.P. Hopstaken, D. Curulla, J.J. Lukkien, P.A.J. Hilbers, and J.W. Niemantsverdriet, "Quantification of lateral repulsion between coadsorbed CO and N on Rh(100) using temperatureprogrammed desorption, low energy electron diffraction and Monte Carlo simulations", J. Chem. Phys., 119, 524-532 (2003).
I.N. Yakovkin and N.V. Petrova, "Microscopic model of CO oxidation on Pt(111)", Surf. Sci.,600, 2600-2607 (2006).
R.M. Ziff, E. Gulari, Y. Barshad, "Kinetic Phase Transitions in an Irreversible Surface-Reaction Model", Phys.Rev.Lett., 56, 2553-2556 (1986).
【系列教程】
Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【1】
Materials Studio官方教程:GULP——拟合力场
Materials Studio官方教程:GULP——计算金刚石的性质
Materials Studio是久负盛名计算模拟软件,问世20余年来,经过不断地迭代优化,使其功能异常强大,极易上手,初学者只需通过简单的参数设置和点击鼠标就能完成DFT计算。其计算可靠性久经考验,备受Nature、Science等顶级期刊认可。
华算科技和Materials Studio官方代理深圳浦华系统联合推出Materials Studio建模、计算、分析课程。课程专为零基础学员设计,沿着理论讲解、模型搭建、性质计算、结果分析层层递进讲解,带你快速入门DFT计算。课程极度注重实操,全程线上直播,提供无限回放,课程群在线答疑。(详情点击下方图片跳转)
识别下方二维码报名,或者联系手机13005427160。

https://blog.sciencenet.cn/blog-2531381-1329428.html
上一篇:Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【2】
下一篇:Materials Studio官方教程:Mesocite——磷脂双分子层的粗颗粒化分子动力学【1】