博文
GCTA学习8 | 计算多性状遗传力和遗传相关
||
。前面的几节中,我们介绍了GCTA计算G矩阵,和单性状遗传力的计算,它本质上就是GBLUP的估计,但是速度快很多。本节我们介绍,两性状遗传力和遗传相关的计算。
1. GCTA计算两性状遗传相关常用参数
1.1 --reml-bivar(必须)
这部分,是使用reml的方法进行估计方差组分。默认的是AI算法,可以使用EM算法。
1.2 --reml-alg 指定迭代方法(非必须)
--reml-alg 0 # AI算法,默认算法 --reml-alg 1 # EM算法
1.3 --grm(必须)
指定GRM矩阵
--grm # 接二进制文件GRM的前缀 --grm-bin # 同上 --grm-gz # 接文本的GRM文件前缀
推荐使用二进制的文件,因为速度快,暂用空间少。
1.4 --covar(非必须)
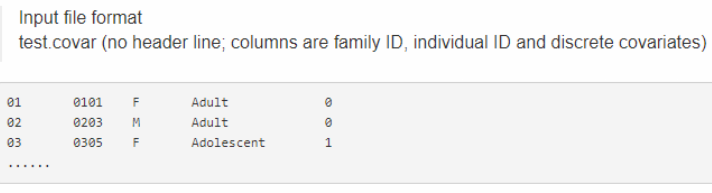
这是接因子协变量的,第一列和第二列分别是FID和IID,后面接因子协变量,比如场年季
1.5 --qcovar(非必须)
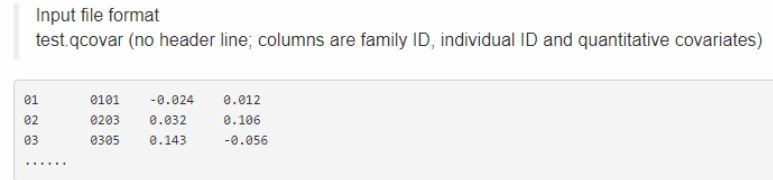
接的是数字协变量,比如PCA,比如初生重等
1.6 --pheno(必须)
接的是表型数据,格式也是plink的格式,第一列FID,第二列IID,第三列是表型数据(缺失用NA表示),第四列是表型数据
2. 数据准备
2.1 表型数据
三列,第一列是FID,第二列IID,第三列是表型数据y,没有行头,空格隔开。
2.2 基因型数据
plink的二进制文件

2.3 协变量
这里,示例数据中,没有提供协变量信息。如果提供,可以按照第一列是FID,第二列是IID,其它是协变量的方法整理数据。协变量分为数字协变量和因子协变量,要分开整理。
3. 构建GRM矩阵
「使用Van的方法」
这里,用Van的方法,类似我们GBLUP估计所用的矩阵构建形式。
gcta64 --bfile ../test --make-grm --make-grm-alg 1 --out g2
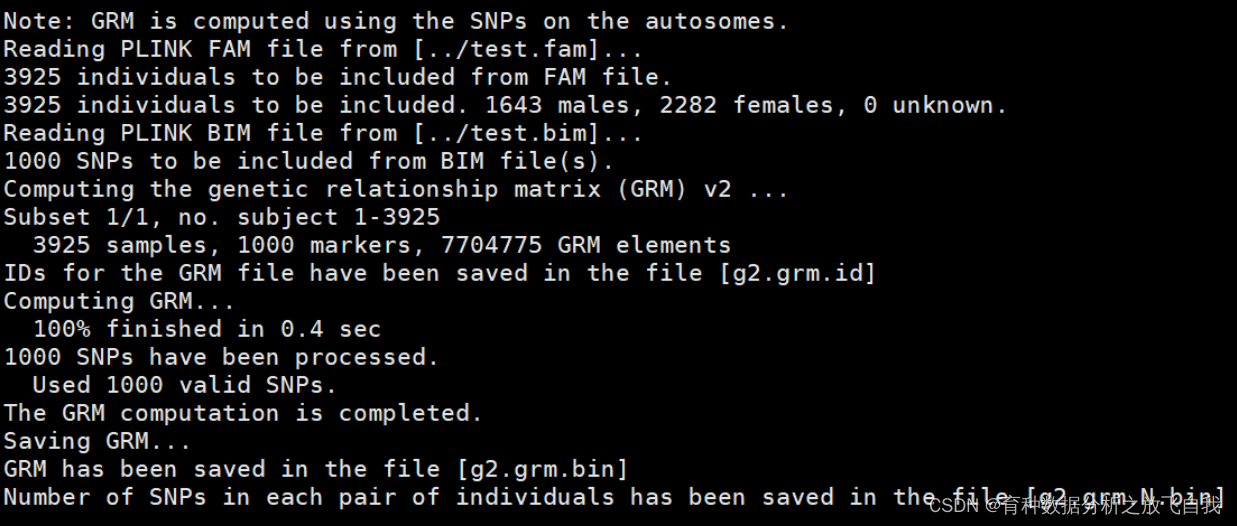
结果文件:
4. 两性状估算遗传力和标准误
这里,已经构建好了GRM矩阵,指定表型数据,进行遗传力的估计
gcta64 --reml-bivar 1 2 --pheno ../nn_pheno.txt --grm ../grm/g1 --threads 30 --out re1
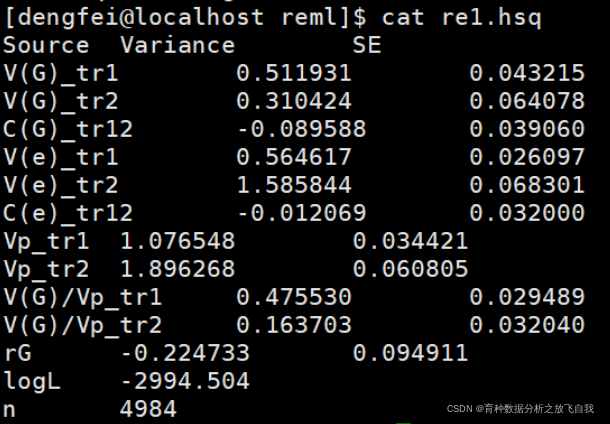
结果可以看出:
Vg11:加性方差组分为:0.51 Vg22:加性方差组分为:0.31 Vg12:加性协方差为:-0.089 Ve11:残差方差组分为:0.56 Ve22:残差方差组分为:1.58 Ve12:残差协方差组分为:-0.012 Vp11:表型方差组分(Vg+Ve),为1.076 Vptr2:表型方差组分(Vg+Ve),为1.89
第一个性状,遗传力为0.47,标准误是0.02 第二个性状,遗传力为0.16,标准误为0.03 遗传相关为:-0.22,标准误为0.094
「使用ASReml作为对比」
「方差组分和遗传力结果:」
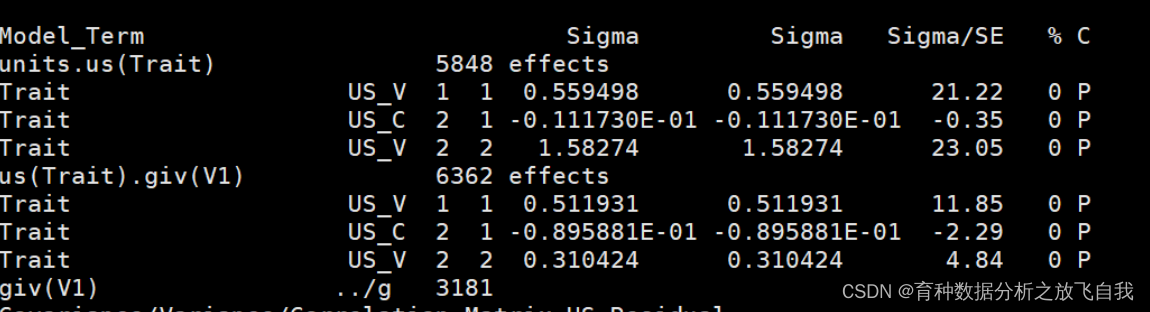
可以看出,结果完全一样。
5. GCTA多性状不支持的参数
「多性状GCTA不支持的部分:」
❝By default, GCTA will take the first two traits in the phenotype file for analysis. The phenotype file is specified by the option --pheno as described in univariate REML analysis. All the options for univariate REML analysis are still valid here except --mpheno, --gxe, --prevalence, --reml-lrt, --reml-no-lrt and --blup-snp. All the input files are in the same format as in univariate REML analysis.
❞
--mpheno --gxe --reml-lrt --reml-no-lrt --blup-snp
6. GCTA的优势和不足
GCTA只能分析两性状的GBLUP遗传评估,计算遗传力和遗传相关,速度很快。
「相对于ASReml软件,缺点如下:」
不支持固定因子缺失 只能是两个性状,3个,以及3个以上不支持 不支持多个随机因子 只能计算遗传相关,不能计算表型相关及标准误
❝欢迎关注我的公众号:
❞育种数据分析之放飞自我。主要分享R语言,Python,育种数据分析,生物统计,数量遗传学,混合线性模型,GWAS和GS相关的知识。
https://blog.sciencenet.cn/blog-2577109-1321917.html
上一篇:GCTA学习7 | 计算单性状遗传力和标准误
下一篇:R语言如何筛选数据框中的列--select