博文
[转载]NG:二倍体黑麦基因组denovo组装20210319(河南农大)
||
A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes
27.60 Q1 NATURE GENETICS
https://doi.org/10.1038/s41588-021-00808-z
Received: 9 December 2019; Accepted: 29 January 2021;
Published: 18 March 2021
部分内容选自详细解读文章(iplant公众号的推送): https://mp.weixin.qq.com/s/vp69tEUovnqJdF2kLcucnw
第一作者:Guangwei, Li Lijian Wang, Jianping Yang, Hang He, Huaibing Jin, Xuming Li,
Tianheng Ren
第一单位:河南农业大学龙子湖校区农学院
通讯作者: Qinghua Yang ✉, Kunpu Zhang ✉ and Daowen Wang
(节选自iplant公众号的推送)
2021年3月18日,河南农业大学农学院植物基因组学和分子育种中心王道文研究员 课题组和杨建平研究员课题组,联合河南农业大学农学院杨青华教授课题组,北京大学邓兴旺院士团队何航副教授课题组,四川农业大学任天恒副教授课题组和百迈客生物科技公司在国际顶级期刊Nature Genetics上发表了题为“A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes”的研究论文,组装出目前麦类基因组组装质量最高的黑麦基因组精细物理图谱,并系统解析了黑麦的演化历史、淀粉合成、储存蛋白、抽穗期及驯化等相关基因的详细机制。同期杂志也背靠背发表了由德国莱布尼茨植物遗传与作物研究所(Leibniz Institute of Plant Genetics and Crop Plant Research,IPK)Nils Stein教授课题组牵头的黑麦基因组国际合作项目的研究成果“Chromosome-scale genome assembly provides insights into rye biology, evolution, and agronomic potential”,发布了欧洲栽培黑麦“Lo7”自交系的基因组组装结果,并解析了黑麦的演化、抗病、抗冷及自交不亲和等相关基因分子机制,建立了黑麦-小麦易位系的高通量鉴定方法,并以此为基础,深度解析了黑麦远缘易位染色体片段对小麦增量的重要作用。这些研究成果,对黑麦和小麦遗传改良具有重要的参考价值,同时也为麦类作物育种改良的源头创新提供了重要的信息资源。
与小麦和大麦一样,中东地区是黑麦的起源和野生遗传资源分布中心。黑麦具有极好的抗寒、抗病、抗逆和耐旱能力,在贫瘠土地条件下也有很强的适应能力,可耐受其他谷物无法耐受的极端气候条件,对肥料及农药需求较少,可给自然条件恶劣地区带来可观的生态和经济效益。
写进高中生物教材的“小黑麦”,是由二粒小麦(AABB)或普通小麦(AABBDD)和黑麦(RR)远缘杂交和加倍人工合成的异源六倍体或八倍体新物种。以鲍文奎院士为代表的我国老一辈科学家也在小黑麦的培育和推广上做出了杰出的贡献。相对于普通小麦和黑麦,小黑麦在生物量和抗性上优势突出,在全球范围内广泛种植(FAOSTAT,2020)。通过远缘杂交,黑麦还能替换掉小麦的染色体片段,产生稳定遗传的小麦品系,为小麦育种导入优异的外缘基因。由黑麦和小麦远缘杂交产生的1BS/1RL易位系,把黑麦的白粉病和条锈病抗性基因导入小麦,显著地提高了小麦的抗病能力和产量,为保障我国和世界粮食安全起到了至关重要的作用。目前,我国小麦品种中大约有50%以上的冬小麦品种均携带有1BL/1RS易位染色体[2]。
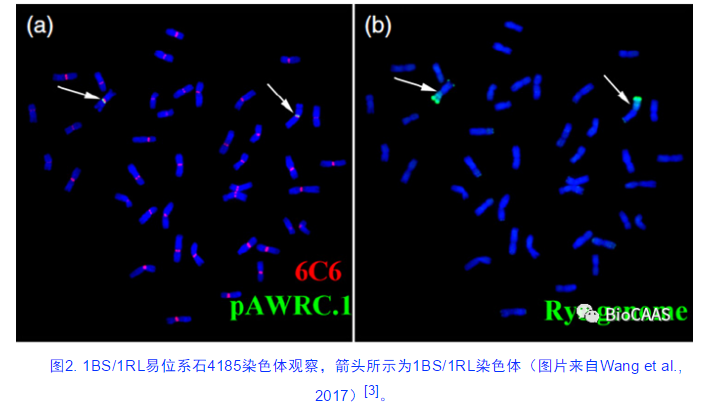
相对于其他麦类二倍体基因组,黑麦基因组更加复杂(2n = 2x = 14, RR),其基因组大小估计为8 G,远大于水稻(25倍)、谷子(20倍),高粱(11倍)、玉米(4倍)、大麦(1.5倍)等其他谷物基因组,并且其基因组中有超过90%为高度重复的转座子序列。自然情况下黑麦具有自交不亲和性,属于异交作物,具有极高的杂合性,这是黑麦基因组组装的又一道很难逾越的障碍。可见,大而复杂性的黑麦基因组严重制约了麦类比较基因组学的研究、重要农艺性状遗传机制解析、作物基因学理论探讨,以及黑麦与小麦等重要作物的遗传育种改良。
为了解决上述科学问题,河南农业大学农学院联合四川农业大学、北京大学和百迈客生物科技公司等多家单位组建了协同创新研究团队,开拓性的利用中国栽培品种“威宁黑麦”强迫自交可部分结实的特性,让其强制自交18代,获得了高度纯合的自交系材料,为基因组组装扫除了部分障碍。研究团队利用高通量二代测序、PacBio单分子测序、染色质三维构象捕获 (Hi-C)、单分子光学图谱(Bionano)和高密度遗传图谱等技术,联合优化多种组装策略,成功的构建了威宁黑麦的高质量精细基因组物理图谱(图3)。
威宁黑麦基因组组装大小为7.84G,其中7.25G(93.67%)序列可以锚定到7条染色体上,其中5条染色体长度超过了1 Gb,最大的染色体长度(2R,1.15 Gb)为水稻基因组的3.6倍。组装得到的物理图谱与已有的欧洲黑麦(Lo7 x Lo225 RILs群体)构建的遗传图谱的相关性可达0.99。在单碱基水平通过比对发现,威宁黑麦的组装准确性可达99.99%,其基因组杂合性为0.26%。威宁黑麦基因组LAI质量参数评分为18.42,与第七版黄金标准组装的水稻参考基因组日本晴的评分近似(21.2)。其BUSCO 值为96.74%。这些结果表明,威宁黑麦基因组物理图谱得到高质量的组装完成。威宁黑麦中一共注释到45,596个高可信度基因,这些基因中有1989个抗病基因,抗病基因的数量高于大麦、水稻和其他麦类二倍体物种。
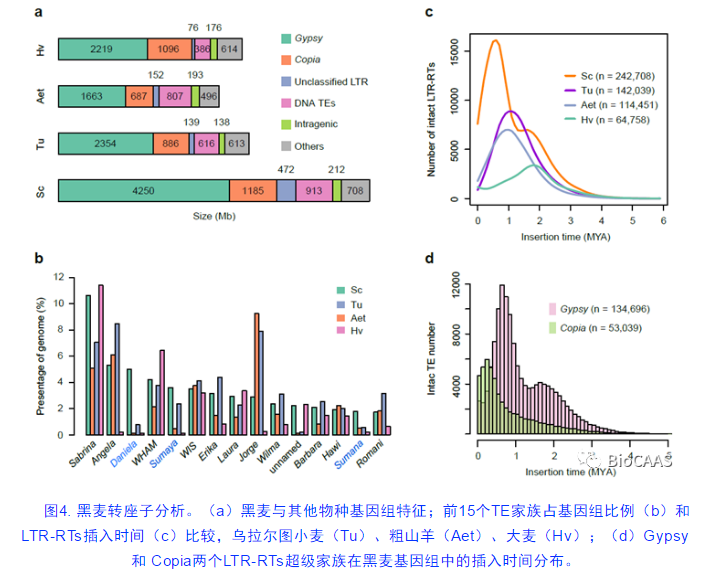
分析发现,黑麦中有6.99 Gb的转座子(transposon elements, TE),可以分为537个TE家族,占基因组比例的90.31%,显著高于其他麦类作物。长末端重复反转录转座子(LTR-RTs)对黑麦基因组扩张的贡献最大。相对于大麦,黑麦多出2.52 Gb的LTR-RTs,贡献了基因组扩张的85.42%(图4a)。黑麦基因组的扩张主要集中在几个重要的TE家族上,比如前15个TE家族约占基因组比例的56.5%,其中Daniela, Sumaya和 Sumana三个家族在黑麦中具有明显的特异性扩张(图4b)。TE演化分析发现,相对于其他二倍体麦类,黑麦中LTR插入时间具有明显的双峰分布,一次大约发生在170万年前,与大麦TE扩张时间相近;更近的一次大约发生在50万年前,与二粒小麦(AABB)形成时间和普通小麦B组祖先消亡时间近似,这是时间上的巧合还是反映了麦类演化史上一次重大的演化事件,还需进一步研究。详细研究发现,双峰分布的时间特征主要是由Gypsy超级家族的反转录转座子引起的,而Copia超级家族爆发时间距离现在更为相近(峰值为30万年)。上述分析首次系统地阐述了TE扩张与黑麦基因组演化历史的关系,如同我们仰望了很久珠穆朗玛峰,这一次我们终于攀上珠峰,亲眼目睹了其芳容。
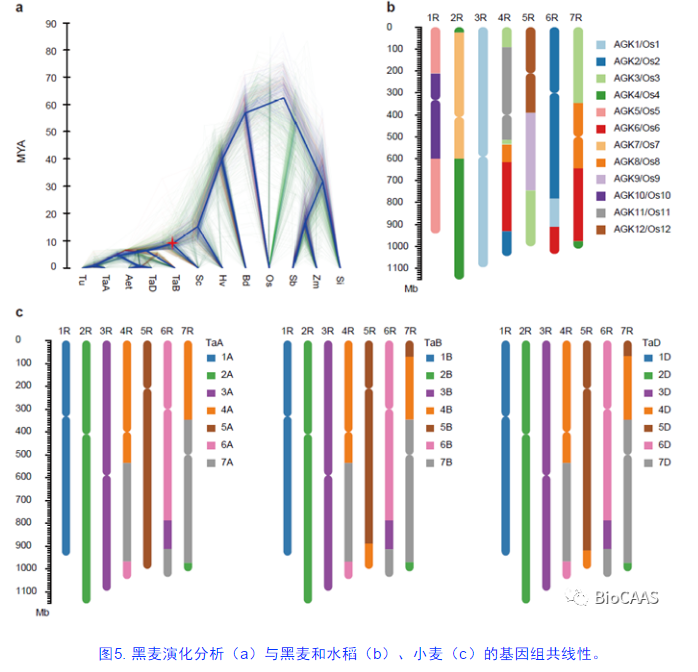
通过与禾本科其他已测序基因组相比,在黑麦中一共鉴定到2,517个单拷贝基因。利用这一信息重新构建了禾本科系统演化史,分析发现黑麦与小麦、大麦共同祖先的分化时间大致分别在960和1,500万年前(图5a)。水稻基因组很好地保持了禾本科物种12条古染色体的基本特征,以水稻基因组为参考基因组分析发现,黑麦中有很多特有的染色体大片段重排易位事件(图5b)。但是,与水稻和大麦类似,黑麦物种的形成过程并没有经历新的全基因组复制过程。本研究得到的黑麦高质量精细基因组物理图谱,是已知组装完成最复杂的二倍体禾本科物种。该项目填补了禾本科基因组的重要空缺,为禾本科基因组演化、麦类比较基因组学的研究、重要农艺性状功能解析等提供了重要的基因组信息资源。
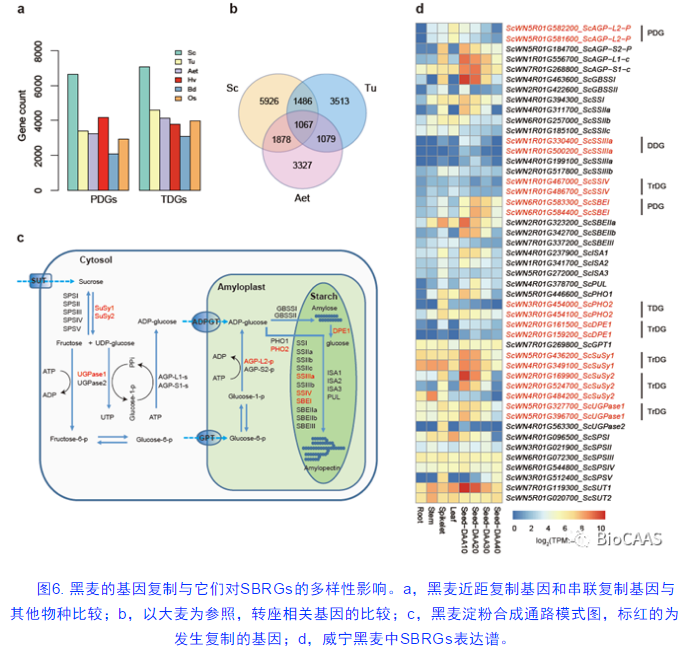
庞大的基因组往往伴随着各种基因复制现象,本研究在威宁黑麦基因组中鉴定到了23,753个分散复制基因(DDGs),6,659个近距复制基因(PDGs),7,077个串联复制基因(TDGs)和1,866个片段复制基因(图6a)。利用和黑麦演化距离最近的大麦基因组为参照,从分散复制基因中鉴定出来10,357个基因复制是由转座子引起的(TrDGs)。有意思的是,在黑麦淀粉通路合成相关基因(SBRGs)中,分别发生了5次转座复制,1次串联复制,1次分散复制和两次近距复制事件(图6d)。尤其是ScSuSy2的两个复制基因之间的表达谱发生了明显的差异,预示着它们可能产生了新的功能分化。这一发现,说明基因复制能为基因新功能分化的产生提供新的资源,从而为物种在不同条件下的适应性演化提供必要基础。这也在一定程度上揭示了黑麦突出的抗性和独特适应能力的遗传基础。
该研究成功解析了黑麦高度复杂的种子储存蛋白位点基因组组成,并以此为基础阐释了这些位点的演化历程,为黑麦、小麦的品质改良和产量提高打下坚实的基础。
苏联著名的遗传学家瓦维洛夫对黑麦进行了大量研究。之前的研究发现栽培黑麦是从它的野生种瓦维洛夫黑麦(S. cereale subsp. vavilovii)中驯化而来,黑麦的驯化可能来自一次必然的“意外”。其最开始可能是小麦和大麦农田中的杂草,它形态上类似于小麦、大麦栽培作物,从而避免被当做“杂草”而移除。这种行为与动物的拟态保护类似,称为“瓦维洛夫拟态(Vavilovian mimicry)”。随后,由于黑麦突出的抗寒、抗病、抗逆和耐旱能力,尤其是在贫瘠土地上出色的产出能力,让它在伴随着大麦和小麦的传播,逐步地得到驯化,从而成为一种特有的麦类作物。
Abstract
Rye is a valuable food and forage crop, an important genetic resource for wheat and triticale improvement and an indispensable material for efficient comparative genomic studies in grasses. Here, we sequenced the genome of Weining rye, an elite Chinese rye variety. The assembled contigs (7.74 Gb) accounted for 98.47% of the estimated genome size (7.86 Gb), with 93.67% of the contigs (7.25 Gb) assigned to seven chromosomes. Repetitive elements constituted 90.31% of the assembled genome. Compared to previously sequenced Triticeae genomes, Daniela, Sumaya and Sumana retrotransposons showed strong expansion in rye. Further analyses of the Weining assembly shed new light on genome-wide gene duplications and their impact on starch biosynthesis genes, physical organization of complex prolamin loci, gene expression features underlying early heading trait and putative domestication-associated chromosomal regions and loci in rye. This genome sequence promises to accelerate genomic and breeding studies in rye and related cereal crops.
Daniela, Sumaya and Sumana retrotransposons 是三个不同家族的反转录转座子
摘 要
黑麦是一种有价值的粮食和饲料作物,是小麦和黑小麦改良的重要遗传资源,也是在 grasses中进行有效的比较基因组研究的必不可少的材料。在这里,我们对中国优良黑麦品种威宁黑麦的基因组进行了测序。组装的assembled contigs(7.74 Gb)占估计的基因组大小(7.86 Gb)的98.47%,其中 the contigs(7.25 Gb)的93.67%分配到了七个染色体上。重复元件占组装基因组的90.31%。与先前测序的小麦基因组,(Daniela),(Sumaya)和(Sumana)逆转座子显示黑麦强烈扩增。威宁黑麦的进一步分析为全基因组基因重复及其对淀粉生物合成基因的影响,复杂的醇溶蛋白基因座的物理组织,早期抽穗特性和潜在的驯化相关的染色体区域以及黑麦中的基因座的基因表达特征提供了新的启示。该基因组序列有望加快黑麦和相关谷物作物的基因组和育种研究。
文章记忆点
The genome of rye is substantially larger than those of barley and diploid wheat species, and was estimated to be around 7.9 Gb, with transposon elements (TEs) constituting approximately 90% of the genome. However, potential contributions of specific TEs to rye genome expansion remain to be resolved.
词句学习
genetic resource:遗传资源
indispensable material:必不可少的材料
To date, 迄今为止,
Rye is well known for its strong tolerance to abiotic stresses and high adaptability to barren soils.黑麦以其对非生物胁迫强的抗性以及对贫瘠土壤的强适应力著称。
Phylogenetic and molecular dating investigations with these genes revealed that the divergence between rye and diploid wheats took place after the separation of barley from wheat, with the divergence times for the two events being approximately 9.6 and 15 Ma, respectively (Fig. 3a).
Through the complementary sets of analyses described above, we generated new insights into the genomic characteristics of rye and its genes involved in agronomic trait control, identifying potentially useful chromosome regions and loci for further studies of the genetic basis of rye domestication. Therefore, the Weining genome assembly is of high value for deciphering rye genome biology, deepening comparative cereal genomic research and accelerating the genetic improvement of rye and related cereal crops.
Method
Rye plants were grown under greenhouse conditions with day and night temperatures of 25 °C and 20 °C and a photoperiod consisting of light for 16 h and dark for 8 h.
1.细胞学实验:FISH(using the fluorescently labeled probes pSC119.2 and (AAC)5 (ref. 53))
2.Genome sequencing:研究团队利用高通量二代测序、PacBio单分子测序、染色质三维构象捕获 (Hi-C)、单分子光学图谱(Bionano)和高密度遗传图谱等技术,联合优化多种组装策略,成功的构建了威宁黑麦的高质量精细基因组物理图谱(图3)。
3.Annotation and analysis of repeats:
RepeatScout, LTR-FINDER, MITE-Hunter and PILER-DF were used for ab initio prediction(从头预测).The identified repeats were compared to those in the Repbase database (version 19.06), followed by classification into different repeat categories using the PASTEClassifier.py script included in REPET version 2.5. The CLARITE program was applied to perform TE annotation by homology57.
The Weining genome assembly was investigated for TEs using RepeatMasker with the TE database ClariTeRep. Next, the CLARITE module was used to correct raw similarity search results to solve the overlap and fragmentation problems of TE predictions and to reconstruct nested TEs.
4.Annotation of protein-coding genes:
The transcripts were assembled, followed by merging and removal of redundancy using
HISAT (version 2.0.4) and StringTie (version 1.2.3).Afterward, the mapped reads were assembled into longer transcripts using Cufflinks software. TransDecoder was then applied to analyze gene structures.
All predicted gene structures were integrated into consensus gene models using
EVidenceModeler (version 1.1.1). These gene models were filtered sequentially to
identify reliable protein-coding genes by (1) removing the CDS with length less
than 300 bp and (2) discarding the CDS that could not be translated because they
lacked an open reading frame or had premature stop codons.The HC gene models were functionally annotated according to the best matches with proteins deposited in GO, KEGG, Swiss-Prot, TrEMBL and a non-redundant protein database using
BLASTP (E value=1×10-5).
5.Phylogeny and divergence time analysis:
Orwas used to identify single-copy orthologous genes conserved in Weining rye and nine other grasses (Os, Bd, Hv, Aet, Tu, Ta, Z. mays, S. bicolor and S. italica). For Ta, the three subgenomes were analyzed separately. All-versus-all BLASTP (E value<1×10-5) was performed, which led to the identification of 2,517 single-copy orthologous genes. MUSCLE was used to perform multiple alignment of deduced protein sequences, followed by construction of gene trees with BEAST version 2.5.1. The gene phylogenies were calibrated using a Bayesian relaxed clock, implemented in BEAST as previously reported58 with two priors: (1) the Bd stem node with a normal-distributed prior (44.4±3.53 Ma) obtained from 17 fossil-calibrated analyses and (2) the Aet stem node with normally distributed calibration in the root of the tree (6.55±0.22 Ma). Subsequently, DensiTree was used to generate a superimposed plot of ultrametric gene trees of the 2,517 orthologous genes. Genome divergence for each pair of diploid species or genomes was estimated based on the distribution of coalescence times of the 2,517 orthologous genes under the multispecies coalescent model.
6.Synteny analysis between Weining rye and rice or common wheat:
To identify syntenic gene blocks between rye and rice, barley or common wheat subgenomes, all-against-all BLASTP (E value<1×10-5, top five matches) was performed for the HC gene sets of each genome pair. Syntenic blocks were defined based on the presence of at least five synteny gene pairs using the MCScanX package with default settings.
7.Analysis of gene duplication:
The ‘duplicate_gene_classifier’ program implemented in the MCScanX package was employed to classify the HC genes located on chromosomes into four categories, whole-genome or segmental,
tandem, proximal or dispersed duplications, based on all-versus-all local BLASTP
(E value<1×10-5, top five matches) within each species. Then the TrDGs were
classified from dispersed duplications using the ‘DupGen_finder’ pipeline (https://
github.com/qiao-xin/DupGen_finder).
8.Search for starch biosynthesis-related gene:The nucleotide sequences for common wheat SBRGs were retrieved from the Chinese Spring genome sequence (version 1.1). They were used to search the Weining genome assembly using BLASTN (E value<1×10-10) to identify rye SBRG orthologs (identity ≥70% and
coverage ≥60%). The normalized counts of SBRG expression were calculated using Illumina RNA-seq da a from root, stem, leaf, spike and developing grain samples with TopHat and Cufflinks. The R package pheatmap was used to display the expression patterns of Weining rye SBRGs in different samples.
9.Expression of heading date-related genes:RT–qPCR assays were performed with the cDNA
10.Investigation of ScFT protein expression and phosphorylation: A polyclonal rabbit antibody specific for ScFT was raised using the peptide QLGRQTVYAPGWRQ, conserved in ScFT1 and ScFT2 (Supplementary Table 24), as described previously59. This antibody was employed to compare ScFT protein accumulation levels in Weining and Jingzhou plants at 4, 7 and 10 DAS by immunoblotting. In brief, total leaf proteins (20μg per sample) were separated using 12% SDS–PAGE, followed by transfer to a PVDF membrane. Subsequently, the membrane was treated with the anti-ScFT antibody (1:2,000 dilution) and then the secondary antibody goat anti-rabbit IgG H&L (IRDye 800CW, 1:5,000 dilution, Abcam), and reaction signals were recorded using the LI-COR 2800. Detection of the HSP90 protein served as a loading control as described previously.
11.Selection sweep analysis:
The variants were identified using BWA with default parameters and SAMtools with the parameter ‘-R -d 1000000 -t DP,AD -Q 20 -q 30 -Bug’. Only the biallelic SNPs with quality scores greater than 50, a minimum allele frequency >0.05, missing data <40% and read depth >4 were retained.
The 127,826 SNPs were also annotated using SnpEff (version 4.2) with Weining gene models, which allowed SNPs to be assigned to intergenic or different genic regions (Supplementary Fig. 6b)
The selective sweeps potentially related to rye domestication were investigated using DRI (πS. vavilovii×πrye-1), FST and XP-CLR, following methods in previous studies61–63. The π and FST values were calculated in 5-Mb windows with 1-Mb steps using VCFtools.
To investigate the genes located in the putative selection sweeps, their orthologs in rice or barley were identified using MCScanX as described above. The functional information for syntenic rice genes was obtained from funRiceGenes (https://funricegenes.github.io/). Analysis of ScID1 is described in the Supplementary Note
通讯作者
** Daowen Wang王道文**
E-mail:: jpyang@henau.edu.cn; yangqh2000@163.com; zkp66@126.com; dwwang@henau.edu.cn

个人简介:
1993年在英国John Innes Center 获博士学位,1997年获得国家杰出青年科学基金。 现任中国科学院遗传发育所分子农业生物学研究中心主任。
研究方向:
https://blog.sciencenet.cn/blog-2885898-1277749.html
上一篇:Syst Biol:系统进化比较分析中的进化样本量和一致性的讨论
下一篇:[Polyploid and Hybrid Genomics]在进化研究中将酵母作为杂交和多倍化研究的模式生物