博文
NML综述:OER催化剂在不同能量标度下的设计策略
|
析氧反应(OER)是电化学能量转换和储存装置中水分解的关键半电池反应。目前最先进的OER催化剂多是贵金属Ir/Ru基材料,目前基准OER催化剂IrO₂的过电位约为300-400mV,仍未达到理想OER催化剂的活性。另外过渡金属氧化物(TMO)的电催化剂因其低成本和在碱性电解质中优异的抗氧化性引起了人们的研究兴趣。然而,目前过渡金属氧化物仍然没有大规模商业应用。因此开发具有高活性高稳定性的非贵金属OER催化剂仍然是一个挑战。
OER的氧化电位很高,高的过电位是阻碍能量储存和转换装置发展的根本原因。OER的高过电位是由于三种含氧中间体(*OH、*O和*OOH)存在制约关系,对所有OER催化剂最少需要施加约0.37V的最小本征过电位。基于这种线性相关关系,人们提出了催化描述符例如d带中心、体相O-2p能带中心,eg占有率、金属-氧共价键、电负性、配位数、最外层电子数等,旨在建立催化剂的结构和催化性能的构效关系,优化活性以达到火山图的顶点。另外,已经有一些研究想要规避这种制约关系对催化活性的限制以进一步提升催化剂的性能。这要求在设计具有优异电催化性能的先进材料和结构之前,必须深入了解材料和性能关系。
Jiangtian Li*
Nano-Micro Letters (2022)14: 112
https://doi.org/10.1007/s40820-022-00857-x
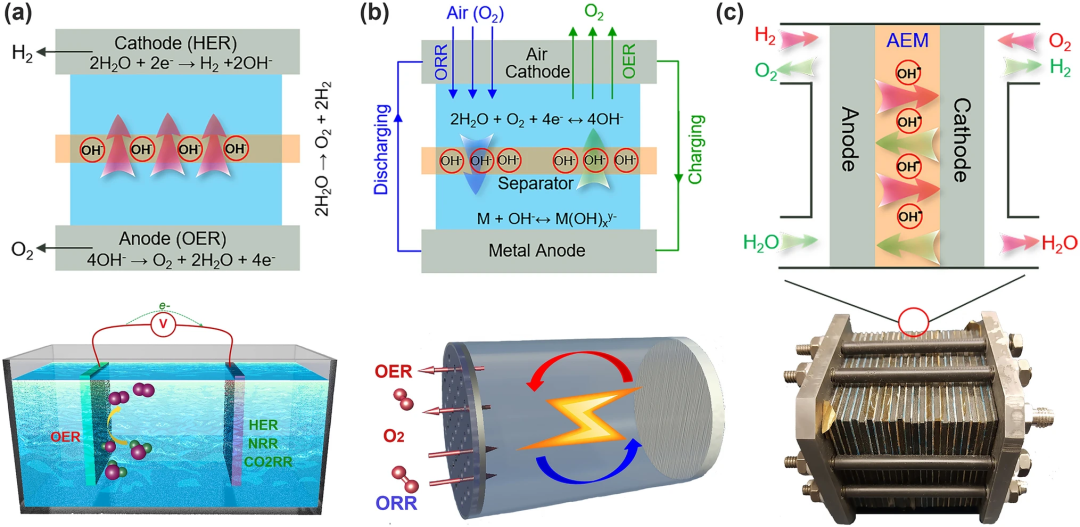
在通过小分子电解产生化学燃料的电解槽中,OER发生在阳极。然而,OER在金属-空气电池的阴极上进行(图1b中的MAB),它的活性和稳定性直接决定了MAB器件的充电和放电性能。只有在大量氧气参与电解的情况下,以氢气产生(电解槽模式)和电力生成(燃料电池模式)两种模式运行的可再生燃料电池能够以一种经济的方式有效地为长期能量存储和能量需求转换电能(图1c)。因此,提高OER效率对于实现基于可再生能源转换和储存的封闭式清洁能源基础设施至关重要。
II 基于吸附物种释放机制(AEM)的OER反应的一般过程
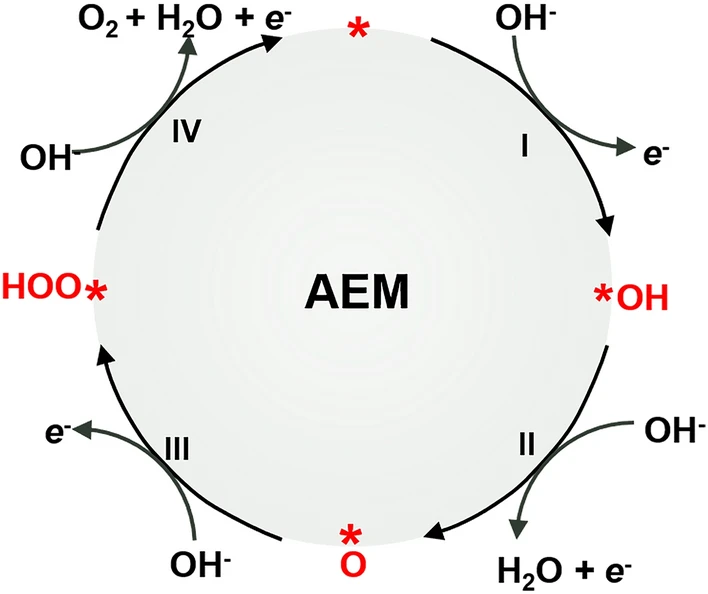
公认的OER过程基于吸附物种释放机制(AEM),通过该机制,含氧吸附质在催化剂的表面活性过渡金属阳离子上发生催化反应。因此,AEM可视为金属阳离子的氧化还原反应。理论上,该过程的标准电极电位Eₒ为0.401 V(vs.SHE), 表观电位E’ₒ为1.229 V(vs.RHE)。然而在实际过程中需要通过多个基本步骤进行,这必然导致外加过电位过高,这种高过电位会导致催化剂的氧化,因此金属氧化物被认为是OER长期稳定性最有效的催化剂。
一般的OER循环过程涉及四个协同的电子转移步骤,分别对应下述四个方程:
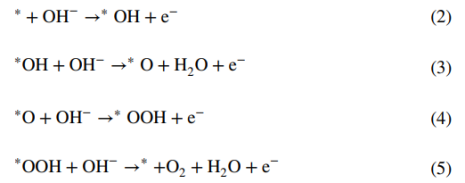
OER首先在活性位点(*)吸附OH⁻以产生*OH自由基(步骤Ⅰ,方程2),自由基去质子化产生*O并伴随一分子水和电子的产生(步骤Ⅱ,方程3),之后OH⁻对*O亲核攻击产生中间体*OOH(步骤III,方程式4)。最后,进一步的质子耦合电子转移过程导致产生一个氧分子以及一个自由活性位点(步骤 IV,方程式 5)。
含氧中间体的吸附、解离和解吸在此过程中起决定性作用,普遍的共识是催化剂表面和吸附物之间的相互作用明确地控制了OER催化活性。因此,系统地了解反应过程中活性位点与含氧中间体之间的相互作用对于开发更有效的OER电催化剂至关重要。
III 催化剂中制约关系对OER反应过电位的影响
在上述电子转移步骤中的三个含氧中间体都通过氧原子吸附在表面上,在这种情况下,*OOH(EHOO*)、*O(EO*)和*OH(EHO*)的结合能是线性相关的。EHOO*/EHO*的斜率大约为1,截距为3.2 eV(图 3a),类似地,EHOO*/EO*和EHO*/EO*的斜率都接近0.5。这种相关的结合能关系普遍存在于金属和金属氧化物表面,与表面的结合强度以及结合位点无关。
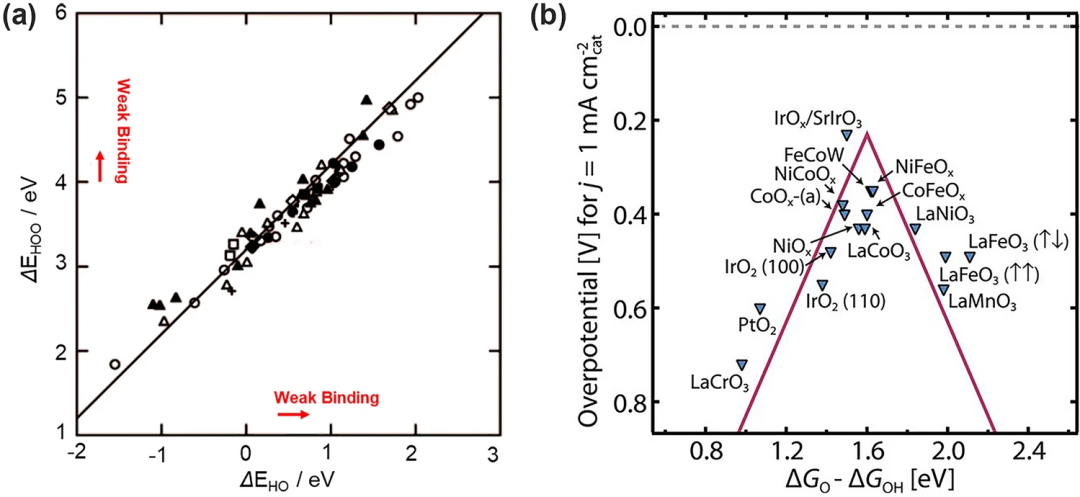
在与氧结合过强的表面上,电位受到*OOH物质形成的限制而对于结合氧太弱的表面,电位受到*OH氧化的限制。这导致了催化活性和氧吸附能之间的火山形的关系(图3b)。位于火山顶部的催化剂拥有合适的氧吸附能因而表现出良好的性能。然而,这种制约关系的存在导致各中间体的吸附能必须受到对方的影响,不能自由调整。比如已建立的制约关系(ΔGOOH=ΔGOH+3.2 V)带来了高达(3.2–2.46)eV/2e⁻=0.37 V的本征过电位。这意味着即使是最好的OER催化剂仍然具有约0.3~0.4 V的最小理论过电位,这在理论上就限制了OER反应的性能发展。
IV d带中心理论
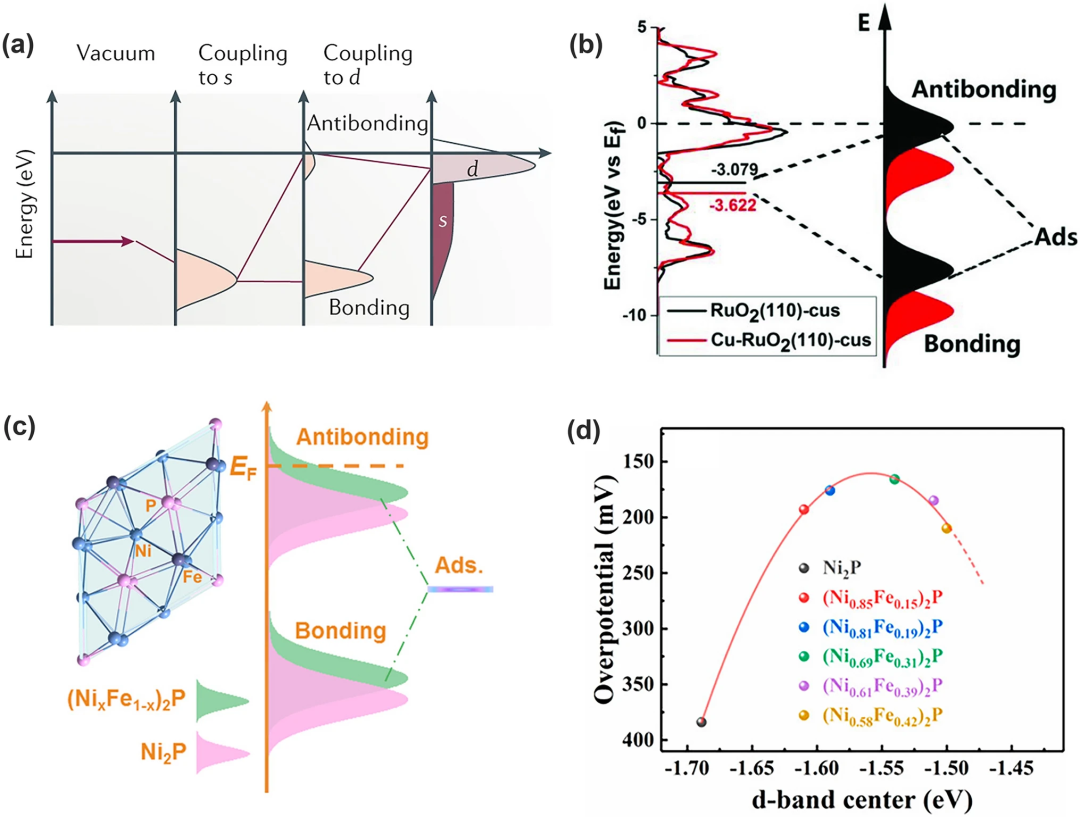
1990年代,Hammer和Nørskov首次利用d带理论建立了过渡金属的电子结构与催化活性之间的关系d带理论。如图4a所示,吸附物在过渡金属表面吸附后,其价态先与过渡金属的s态耦合然后变宽并向下移动,之后继续与d态相互作用形成一个填充的成键轨道与部分填充的反键轨道。反键轨道的填充情况由它与费米能级EF的相对距离决定:d态相对于EF的能量越高,反键轨道的能量越高,键合就越强,反之则越弱。
因此需要电催化剂的最佳Ed能级以实现OER活性最大化。其中掺杂过渡金属基化合物是影响d态的最有效途径,例如将Cu掺杂到RuO₂晶格中会使d带中心远离EF,导致反键轨道能量降低,Ru-O键强度相应减弱(图4b)。相反,Ni 2p的低Ed能级(即弱吸附能)使含氧中间体难以吸附在表面上。再比如Chen 和同事发现,Fe掺杂可以提高Ed能级更接近EF(图 4c),从而提升反键能态并加强吸附物与催化剂表面之间的相互作用,从而增强吸附能力OER过程中的中间体。值得注意的是,我们还可以通过改变Fe浓度,从而改变d带中心的能态获得位于火山顶的良好OER 性能的催化剂(图4d)。
V 体相O-2p中心理论
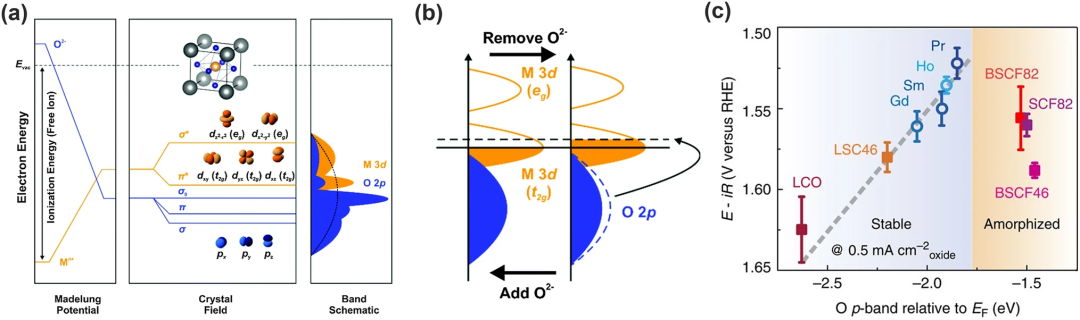
在八面体配位的场中,d轨道分裂成较高能级的两个eg轨道和较低能级的三个t2g轨道(如图5a)。eg双键与O-2p轨道有很强的重叠并产生σ⁻成键轨道和σ*⁻反键轨道。然而,t2g三重态显示出与O-2p轨道的弱重叠并形成π⁻键和π*⁻反键。由于晶胞的平移对称性,分子轨道成为氧化物晶体中的带,表示为M-d带和O-2p带。
体相O-2p带中心与钙钛矿中的氧表面动力学和空位形成能密切相关,能解释在Ruddlesden-Pop过氧化物和多晶钙钛矿材料中所有氧点缺陷的能量学和动力学问题。可以通过刚性带模型来理解(图5b):晶格中脱氧相当于将电子从O-2p带移动到费米能级,使其能量增加;加氧则反之。因此O-2p带的能量反映了不同氧化物费米能级的大小,因为它的能量很大程度上取决于马德隆电位以及氧电子亲和力。
Grimaud及其同事将一些双钙钛矿的高稳定性与活性(图5c)归因于它们的O-2p带离费米能级既不太远又不太近。他们发现将O-2p靠近费米能级会增强OER活性,但太近会引起表面非晶化使稳定性降低。这是因为高O-2p带带来的低氧空位形成能增加了氧空位的浓度加速了表面氧交换并促进了晶格氧的迁移,才形成了表面非晶化。
VI eg占据率
1970 年代,Matsumoto等发现钙钛矿氧化物对氧还原反应的催化活性受TM的eg轨道和氧吸附物的p轨道重叠的影响,重叠越大,电催化活性越高;Suntivich和他的同事研究了超过10种钙钛矿氧化物的OER活性对eg填充程度的影响,并给出了它们之间的火山型图(图6a)。eg填充程度这一描述符的提出是基于eg轨道的σ相互作用强于t2g轨道的弱π相互作用这一事实,eg轨道的填充率接近1时OER活性最佳。该模型仅仅考虑eg轨道上的电子,与之前的d带理论不同。但如果氧化物有相似的eg填充态,则需要通过另外的描述符例如电负性等判断活性大小。
该模型可应用于钙钛矿、尖晶石氧化物、莫来石以及Co₂YZ 型 Heusler 化合物。Xu和同事研究了一系列用于OER和氧还原反应(ORR)电解的MnCo₂O₄ 尖晶石,并确定了八面体配位的 Mn 作为活性位点。OER/ORR 活性(施加电位25μA cm⁻2ox)与eg在八面体位置填充Mn之间的相关性给出了火山图(图 6b),峰位于≈+3 的Mn价态(即t2g3eg1),这巩固了电子轨道填充在用于氧电解的金属氧化物催化剂中的决定性作用;Tüysüz及其同事证明eg轨道中Co活性位点填充可以通过改变Co₂YZ化合物中Y和Z位点来调节,较高的催化电流可以在eg轨道填充率接近1时达到。这进一步支持了eg轨道填充模型对调节M-O键合强度朝向更活跃的OER催化剂的有效性。
eg轨道的填充情况可以通过d电子的数量和电子自旋态调节,同时它受到洪特交换ΔEₓ与晶体场分裂能ΔCF的大小关系决定。Wu及其同事展示了一种自旋态调节方法,通过控制LaCoO₃薄膜的晶格取向来优化OER活性。不同晶格取向的LaCoO₃薄膜带来不同程度的CoO₆八面体畸变(即应变),导致钴从低自旋态(LS, t2g⁶ eg⁰)到中自旋状态(IS, t2g⁵eg1),从而具有更好的OER性能(图6d);作者也通过双交换作用改变了NiCo₂O₄尖晶石中自旋态(图6e),通过构建共价纳米异质结和产生氧空位(VO)的共同作用使得Ni和Co的自旋态改变,达到接近1的占有率;Zhou及其同事研究了含有Co-Mn的尖晶石氧化物ZnCoₓMn₂₋ₓO₄ (x=0.0-2.0),发现由于CoO₆和MnO₆之间的超交换效应,可以通过改变Mn/Co比来调节活性Mn阳离子的eg占有率(图6f); Guo及其同事发现,通过直接氢处理在Yb掺杂的 CaMnO₃中引入氧空位导致例如Mn填充的增加和电导率的提高。
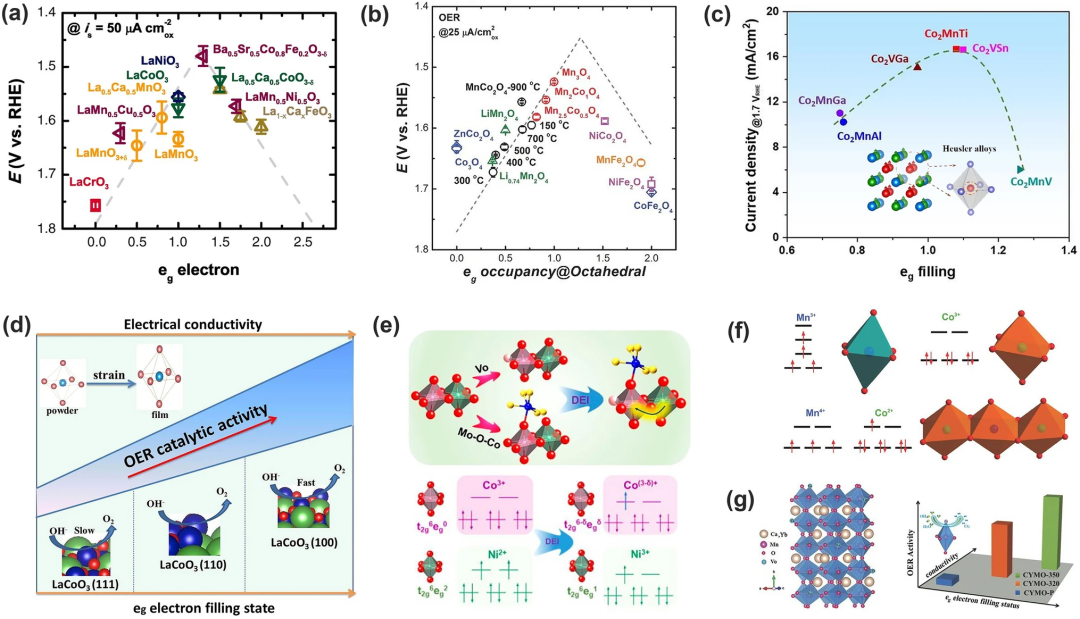
图6.eg占有率与TMOs的OER活性之间的关系以及调控eg轨道占有率的策略。OER活性是eg在八面体位点上的活性中心占据情况的函数:(a)钙钛矿氧化物,(b)尖晶石氧化物,(c)豪斯勒合金;(d)OER活性与不同取向的LCO薄膜的自旋构型之间的关系;(e)Co和Ni在尖晶石八面体位点的双重交换相互作用和eg占有率的转变;(f)通过超交换相互作用在含钴锰尖晶石中实现锰和钴之间的电荷再分配;(g)氧空位和掺杂导致钙钛矿CaMnO₃中的电导率增加;eg的电子填充状态。
VII 金属-氧共价键
eg描述符是在离子模型上建立的,无法有效地解释有关金属-氧共价键或沿金属-氧键地电子共享。因此Shao-Horn及其同事因此引入了金属-氧共价键(电荷转移能)理论。金属-氧共价键已被证明会影响催化活性。它与实验OER活性、表面交换活性(如氧空位形成能、氧结合能和与OER相关的电子转移势垒)以及在碱性溶液中的稳定性呈线性相关(图7a)。至于稳定性,共价键能与氧空位形成之间存在正相关关系,但氧空位形成的驱动力过高会导致钙钛矿相的结构破坏(图7b)。
低价金属的存在会降低过渡金属的3d能级,从而增强金属-氧共价键的强度(图7c)。基于此,Yagi及其同事报道了具有高OER活性的Fe⁴⁺基四重钙钛矿CaCu₃Fe₄O₁₂。其中铁离子的低3d能级增强了金属-氧共价键;八面体配为中心Cu (Fe) eg和O-2p轨道之间的大重叠提高了其结构稳定性;多个Cu2⁺和Fe⁴⁺增强了其寿命以及OER活性;该理论还可应用在尖晶石、岩盐氧化物、Ruddlesden-Popper氧化物等其他金属氧化物。Xu等人对尖晶石ZnFeₓCo₂₋ₓO₄氧化物的取向与OER活性的关系进行了研究,发现Fe的掺杂促进了Zn空位和Co⁴⁺的形成,而Co⁴⁺具有低能量3d态,并且由于Co-3d和O-2p轨道之间的能量距离缩短,即Co-O共价键增强,产生更高的杂化,从而增强了OER的活性(图7d-e)。
与O-2p模型不同,金属-氧共价模型关注的是金属的3d态和O的2p态之间的能量差,即电荷转移能量(Δ),其在调节氧化物的电子特性方面起着显著的作用,从而影响OER动力学和机制。Hong等曾发现,降低电荷转移能量可以大大提高OER活性。图7f展示了PBCO相对LaCrO₃较低的Δ具有较高的电导率和OER活性;图7g展示的是Sr掺杂LaCrO₃ (La₁₋ₓSrₓCoO₃)后提高了电导率,这是因为Sr的掺杂使Co-O-Co键的重新排列和Co阳离子的氧化,扩大了满O-2p价带和空Co-3d导带之间的重叠,降低了电荷转移能。
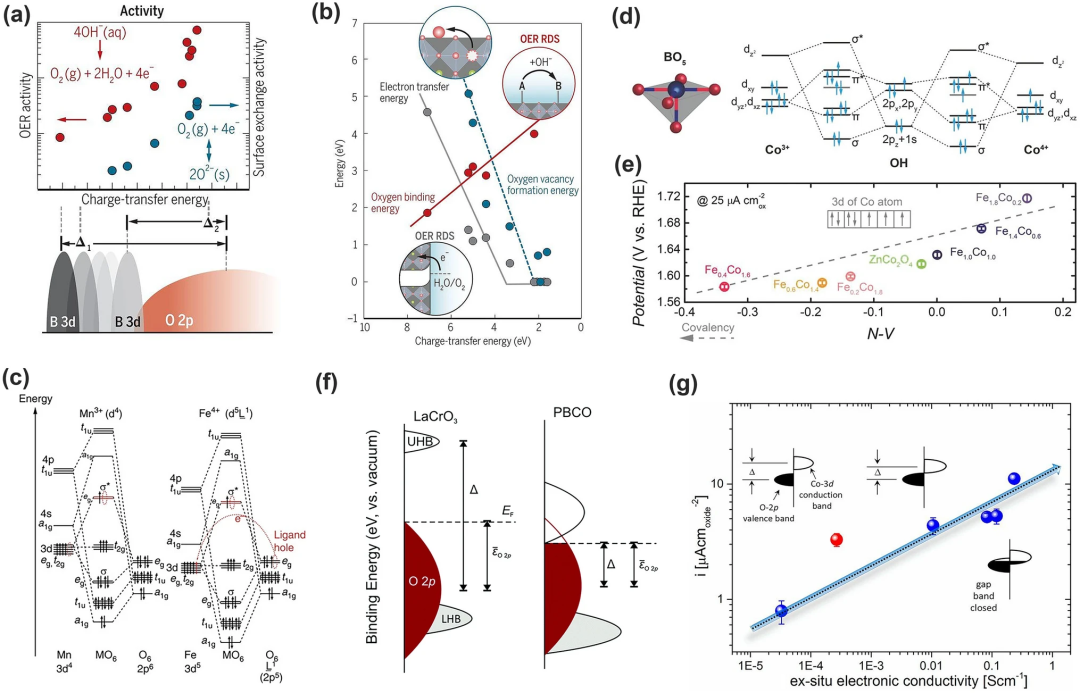
图7. 金属-氧共价键能与OER活性之间的相关性。(a)电荷转移能量与 OER 活性(红色)、表面交换活性(蓝色)和 3d 过渡金属中的 OER 活性位点特性之间的相关性;(b) 电荷转移能量与相关能量学和决速步的关系:氧空位形成能(蓝色)、氧结合能(红色)和电子转移能(灰色);(c)常规Mn3⁺O₆ 和 Fe⁴⁺O₆八面体的分子轨道示意图。Mn3⁺和Fe⁴⁺离子的3d轨道能级分别高于和低于O-2p轨道的能级;(d)尖晶石氧化物表面的Co-OH(Co3⁺和Co⁴⁺)键合的分子轨道图;(e)OER活性作为N-V参数的函数(f)电荷转移能量示意图:TM-3d和O-2p 价电子态的相对能量;(g)电流密度(μA−2 cmoxide)作为La₁₋ₓSrₓCoO₃ 系列的非原位电子电导率的函数;红色圆圈代表SrCoO₂.₅。
VIII 其他描述符
(1)外层电子数:OER中间产物在过渡金属及其氧化物上的吸附能的变化趋势可由外部电子的数量反映(如图8a)。注意此处的外层电子值得是金属原子氧化时保留的价电子数。根据该描述符即可构建预测吸附能量网络并解释过渡金属与其氧化物存在的制约关系。
(2)本体热化学。本体热化学一直被用来描述氧化物表面催化活性的趋势,然而Calle-Vallejo及其同事后来发现,体热化学与表面吸附能量学的制约变化与氧化物中过渡金属的外电子数变化相似并且该结论适用于大量的过渡金属。因此可建立起过渡金属本体与表面性质的制约关系,构建出火山图(图8b)。
(3)O-2p态的平均能量。图8c显示了各种金属和金属氧化物中氧原子的平均O-2p态能量和(ΔEO-ΔEOH)之间的相关性。能看出具有较高2p态的结合氧原子具有较高的(ΔEO-ΔEOH),对结合氢的亲和力更强。
(4)参与配位的不饱和金属阳离子。Tao等将配位不饱和金属阳离子 (MCUS) 确定为过渡金属氧化物的OER表面反应性能描述符。TMO 的表面反应性随着MCUS的密度单调增加,提高了弱结合TMO的催化活性(图8d中的右半部分),但削弱了强结合TMO的催化活性(图8d中的左半部分)该描述符的微观解释为相对于EF的最高占据d能级被用作了表面反应性能的电子结构描述符,其能量决定了吸附物在过渡金属氧化物上的结合强度。
描述符的选取应综合考虑,以上介绍的各个描述符都有自己的适用领域以及限制,在筛选设计高活性的催化剂时应该考虑多个描述符的协同作用。
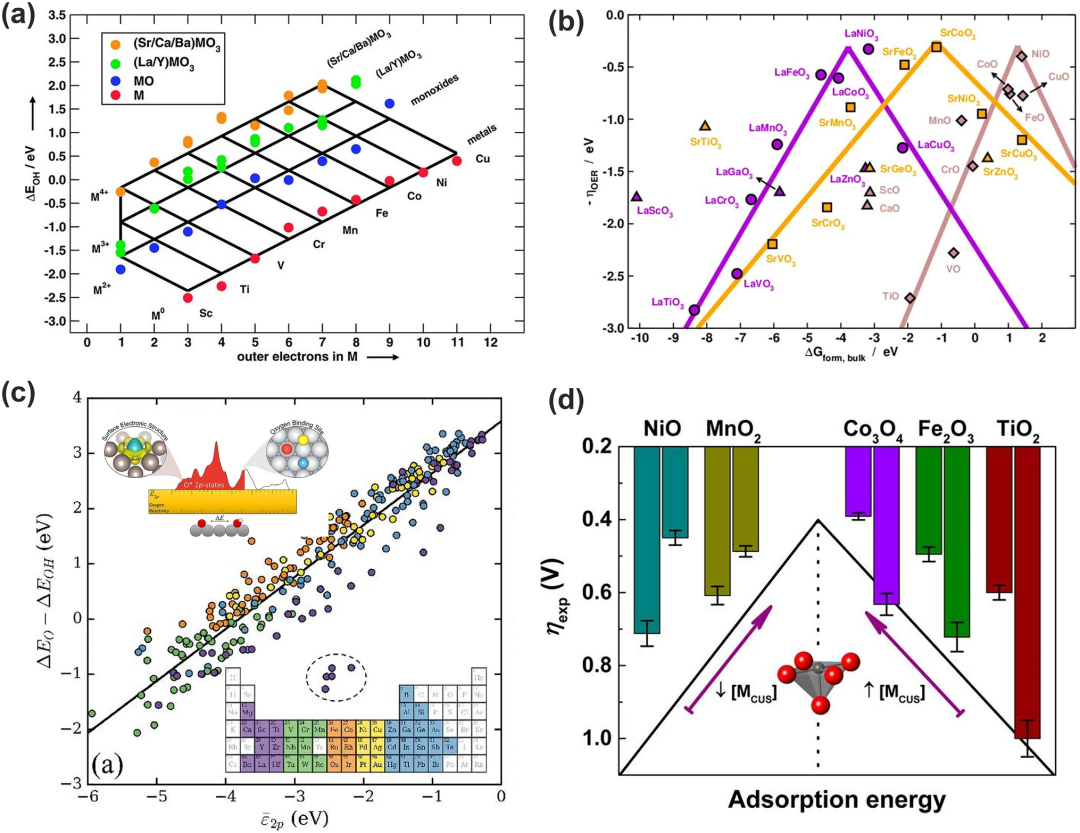
图8. 与氧电解催化活性相关的描述符。(a)*OH 在金属(红色)、一氧化碳(蓝色)、La/Y钙钛矿(绿色)和 Sr/Ca/Ba 钙钛矿(橙色)上的吸附能量网格,作为外电子数量的函数;(b)LaMO₃(紫色圆圈)、SrMO₃(橙色方块)和 MO(棕色菱形)的火山型曲线图;(c)2p轨道与吸附在各种金属和金属氧化物表面的氧原子的反应性之间的相关性;(d)TMO 中表面 MCUS 与过电位的关系;Fe₂O₃的ηexp是在1mA cm⁻2的电流密度,而其余TMO对应的电流密度为10mA cm⁻2。
上述内容均是基于催化当中吸附能的强制约关系,无论是基本描述符的介绍还是OER的一般反应过程均受到该制约关系的影响,为了规避这种关系带来的本征过电位,下面介绍的内容是尝试打破这种制约关系的一些工作和尝试。为打破OH*和OOH*的结合能之间的稳健标度关系,一个基本想法是设计有选择地稳定后者的活性位点。从本质上讲,这归结为加强表面氧对羟基的亲和力而不影响其对原子氢的亲和力。
最近的进展主要包括:(1)构建多个功能中心,对不同的中间体具有选择性活性,以独立优化结合能;(2) 应用应变、纳米级限制和磁场等外部物理效应对某些中间体进行单独调谐;(3)晶格氧参与OER机制。下面仔细地介绍上述方法。
1. 构建多个功能中心
(1)图9a展示的是O-O键直接耦合的路径。两个具有适当距离的相邻金属中心发生 *OH 去质子化产生两种金属-氧类物质,然后它们将耦合释放O₂分子,这种机理也可应用在OER的多相催化中。例如Yagi 等就成功在钙钛矿 CaCu₃Fe₄O₁₂ 中直接形成 O-O(图9b),他们在其中掺入Fe⁴⁺ 和 Cu2⁺ 后,Fe-O-Fe 键被严重弯曲,缩短了约 2.6 Å 的氧-氧距离,直接形成了 O-O键;Lin 等也报道了一种在α-MnO₂上用钌原子阵列贴片的OER电催化剂,Ru/MnO₂(2.9 Å)中的原子间 Ru-Ru距离比RuO₂ (3.1 Å) 中的短,有利于 O₂ 释放的直接 O-O 自由基耦合(图 9c)。该机理被称为多相催化的氧化物路径机制(OPM),中间体仅含O* 和 HO*,跳过了去质子形成过氧化物的过程,因此可以打破制约关系。但该机制的挑战在于确定金属活性位点的几何结构。
(2)构建异质结构实现在不同的位点分离*OH 和 *OOH,Gao等人在NiO/NiFe 层状双氢氧化物异质结构中实现了这一策略(图9d),该过程的基本原理如图9e所示,通过对中间体施加不同的化学键使得每个中间体的结合能可以独立调整;杂原子结构也可调节活性中心的吸附性能。Fei等报道的单原子催化剂MN₄C₄(M=Fe、Co、Ni)可通过调节M的种类改变各原子对中间体吸附的强度以做到分离中间体(图9f、g)。
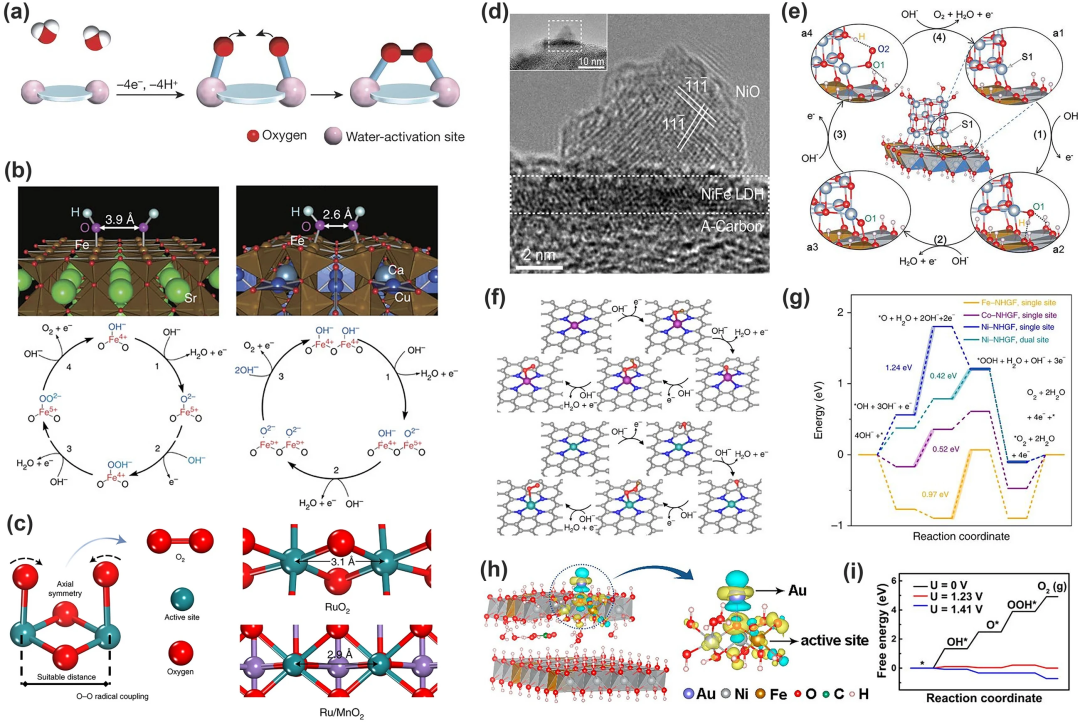
通过改变催化剂的外部环境来改变其催化性能也是规避制约关系的一种方法,具体包括三种方法:晶格应变、纳米约束以及磁场变化。
(1)晶格应变:通过外力改变表面原子间距来改变表面电子结构以改变中间体的被吸附强度。Wang等通过计算表明ΔGOH和ΔGOOH的变化幅度相同,而ΔGO变化幅度更大;Xie等用N掺杂的石墨烯从理论上证明拉伸应变倾向于破坏N-C*键,增强O*的吸附;Khorshidi等还建立了应力模型解释应变对吸附质-催化剂键合的影响,从而破坏制约关系(图10a)。
(2)纳米环境约束:Doyle等假设3D纳米级通道能够提供一个封闭的反应环境,使中间体和催化剂之间能够进行选择性相互作用(图10b)并通过改变金红石的OER环境使EOOH发生了明显变化,在RuO₂ (dmm≈ 7 Å)时过电位大约200 mV,打破了制约关系的限制。
(3)外部磁场的改变:磁场会影响反应路径,与催化剂的磁性成正比。Ren等指出,在OER的自旋极化过程中,第一个电子转移步骤为RDS,但在磁场的干预下,铁磁催化剂和吸附的氧物质之间发生相干加速反应使其不再是RDS;Zhou等报道了由CoMn金属-有机框架 (MOF) 中的局部磁加热产生的热微分超晶格,发现了局部加热导致了自选电子的重排,揭示了一种新的通过操纵磁性氧化物中的自旋极化来加速OER的路径。
(4)光致析氧:利用光热效应辅助OER。Lin等利用光热效应提高了NiFe₂O₄纳米颗粒的OER活性,证明光热效应不仅能加快OER反应动力学,还能降低催化活性物质的生成能垒;并且光生电荷空穴也有助于提升OER活性,例如Ye等报道了Au纳米颗粒修饰的Ni(OH)₂纳米片用于OER催化,光生空穴促进了Ni2⁺的氧化提高了OER的活性(图10g)。
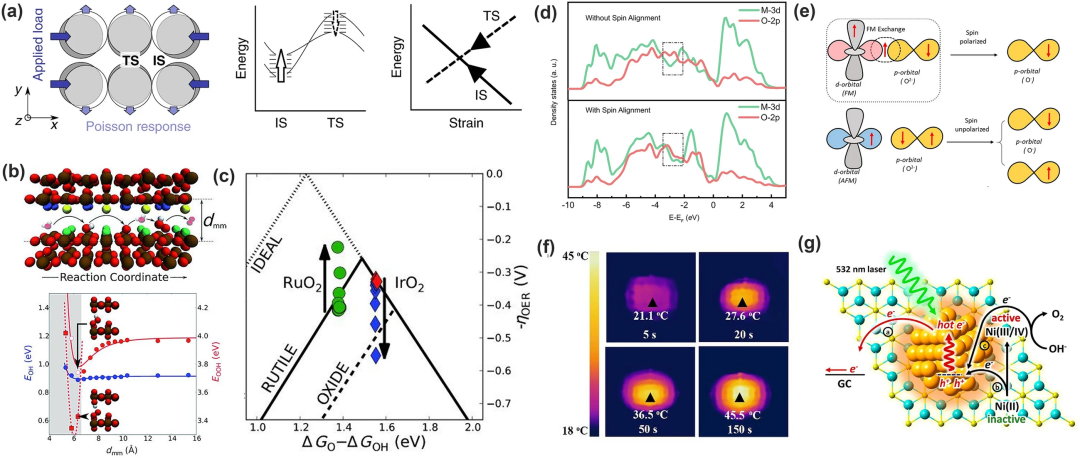
3. 晶格氧传导机制 (LOM)
该机制指晶格中的活跃的氧从晶格中溢出与吸附的氧(*O)相互作用参与OER循环。但晶格氧在热力学上不利,会引发LOM,需要晶格的EF移动到O-2p带并位于O₂/H₂O对的氧化还原能量之上。如图11a,M-3d和O-2p带之间的重叠增加,带心能隙减小有利于晶格氧转移。Meford等通过将Sr2⁺取代引入La₁₋ₓSrₓCoO₃₋δ和Ruddlesden-Popper氧化物La₀.₅Sr₁.₅Ni₁₋ₓFeₓO4±δ,成功的观察到了晶格氧的参与(图11b、c);Grimaud等用原位1⁸O同位素标记质量DEMS实现了晶格氧参与OER过程的直接实验观察并进一步证实当EF附近的电子态填充到高共价氧化物的O-2p轨道时,OER可以切换到LOM。Huang等将低价且无催化活性的Zn2⁺掺入CoOOH中,以产生非键合氧(ONB)态并同时增加Co-O共价键来启动LOM(图11d),结果表明只有当两个相邻的O-O可以杂化它们的氧空穴而不牺牲金属-氧的杂化时,OER才会通过LOM路径进行。Sun等研究了300多种尖晶石氧化物并根据计算作出了ε-2p和ΔO₂p-M3d的关系图,右上角为LOM机制,左下角为AEM机制,因此ε-2p和ΔO₂p-M3d共同调节OER的反应机制(图11e)。另外作者还给出了OER活性与四面体/八面体阳离子和氧之间的共价键能Max(DT, Dₒ)的火山形关系图(图11f),实验证明位于火山顶部的[Mn]T[Al₀.₅Mn₁.₅]OO₄的过电位仅为240mv,活性与最先进的催化剂相当(图11g)。
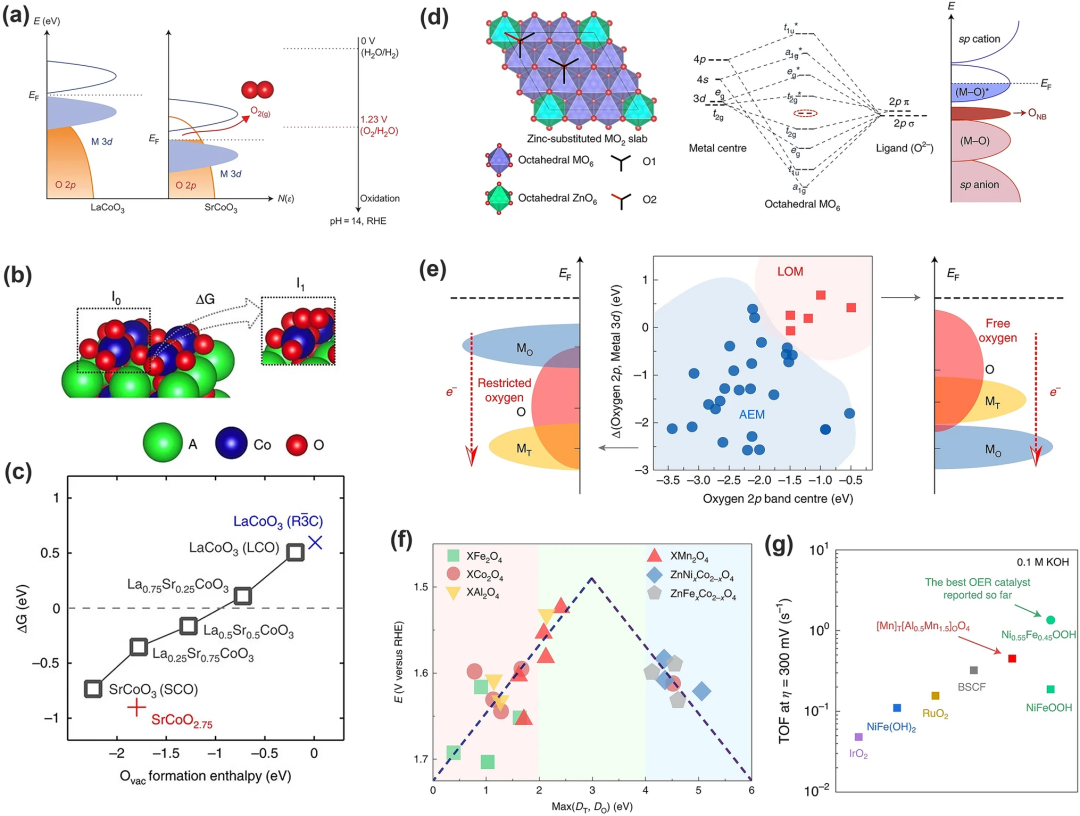
4. LOM 中的反应过程
在基于DFT计算的典型LOM催化循环中(图12),*OH在O⁻位点上脱氢产生*OO物质和VO(步骤I,方程式6),然后*OO物质演变回*OH,同时释放O₂和电子(步骤II,方程式7)。在此步骤中,VO被*OH重新占据,相邻的表面晶格氧被质子化(步骤III,方程式8)。最后,*OH在去质子化过程中再生。
LOM 机制消除了传统AEM中发生的协同质子电子转移步骤。它通过O-O键的直接耦合(方程式7)产生氧气,并且不涉及*OOH 的形成,成功打破AEM中制约关系。
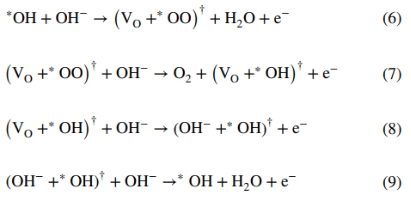
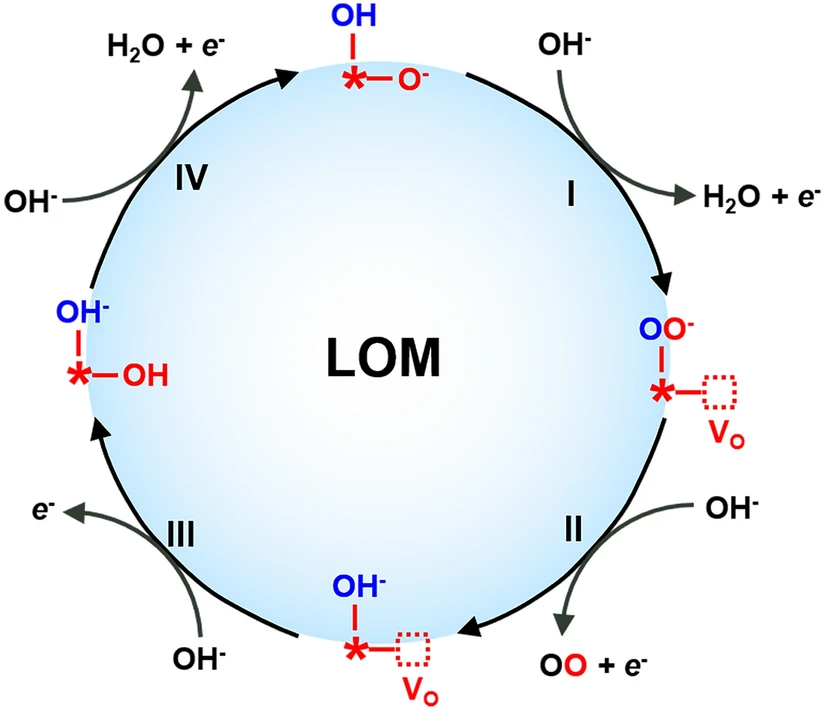
5. 实验观察证实LOM机制
(1)原位1⁸O标记的电化学质谱(EMS)。在OER期间用标记的催化剂或监测反应后的电解质。Grimaud及其同事对1⁸O标记的钙钛矿进行了在线电化学质谱(OLEMS)测量,并首先提供了高共价氧化物(La₀.₅Sr₀.₅CoO₃₋δ和LaCoO₃₋δ)中晶格氧氧化的直接实验证据。
(2)光谱。操作光谱技术包括红外(IR)光谱、拉曼光谱、X射线吸收光谱(XAS)和X射线光电子能谱(XPS),已成为研究电催化剂的有力工具。原位使用这些技术来探测反应条件下的催化过程,可以提供有关电极表面上发生的动态变化的准确信息,并更深入地了解基本的反应机理。Lin等利用原位同步辐射红外(FTIR)光谱检测到在1089 cm⁻1处形成O-O键;Hu等发现在1⁸O同位素标记下,在~26 cm⁻1处存在频率变化,这归结于Co-O A1g 振动和晶格氧与电解质交换。
(3)不同pH的OER电化学活性:Grimaud等发现能够进行LOM机制的高价氧化物的OER活性受到pH的影响,但通过AEM机制的内在OER活性与pH无关(图13a)。这是由于在OER过程中存在不协调的质子-电子转移步骤,源于氧化物/电解质界面处的电子转移动力学和氢亲和性之间的不匹配。在OER与LOM同时存在的情况下,电荷转移能量的显著降低加速了电子转移并削弱了氢氧化物的亲和力,使它们在具有pH依赖性的PDS中解耦。但需注意通过LOM的OER可以表现出pH依赖性活性,但pH依赖性活性并不是LOM反应的决定性指标。
(4)表面非晶化:由于LOM机制需要晶格氧的参与,因此氧离子扩散速率(DO)对于提升OER的性能至关重要。Mefford等发现,OER的催化活性与La₁₋ₓSrₓCoO₃₋δ钙钛矿中的氧扩散速率密切相关,呈线性关系(图13b)且通过LOM路径的SCO要比AEM路径快许多(图13c)。因此Pan等人提出,晶格氧析出后的表面氧空位可以通过电催化剂本体的氧离子迅速扩散补充(图13d),因此增加氧离子扩散速率能提高OER催化性能。

本文通讯作者
主要从事于电化学能源领域,如:电池、超级电容器、太阳能电池、光电化学电池、水分解、二氧化碳减排、生物燃料和燃料电池等。
▍个人简介
▍Email: jiangtian.li.ctr@army.mil

Tel: 021-34207624
https://blog.sciencenet.cn/blog-3411509-1345545.html
上一篇:新加坡NUS综述:可替代锂电池的高能电池之研究及商业化进程
下一篇:沸石保护层改性锌碘电池:长循环寿命、高库伦效率、高容量