博文
多基因联合分析简明图解之PhyloSuite篇
|||
絮语:
虽然QQ空间已经写了两篇多基因联合分析的教程,随着新工具PhyloSuite的开发出炉,只需简单几步就可以完成多基因联合分析,本文以简明图解应用PhyloSuite进行多基因联合分析。
关键词:PhyloSuite, PartitionFinder, MrBayes,数据分区,批量重命名树名称
数据准备:
(1)比对好的序列文件,每个基因序列的文件名称建议以GenBank 登录号作为名称,如下图示例,01_ITS_aln.fas:
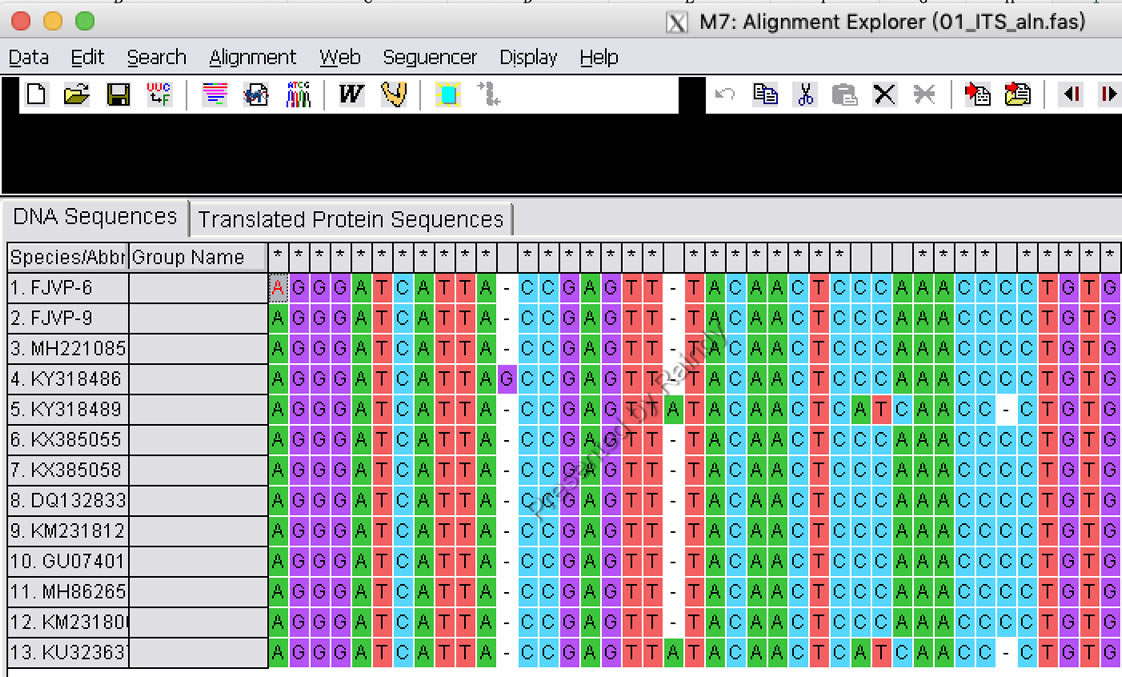
Raindy注:编码蛋白的基因和非编码基因比对策略不同,需要注意
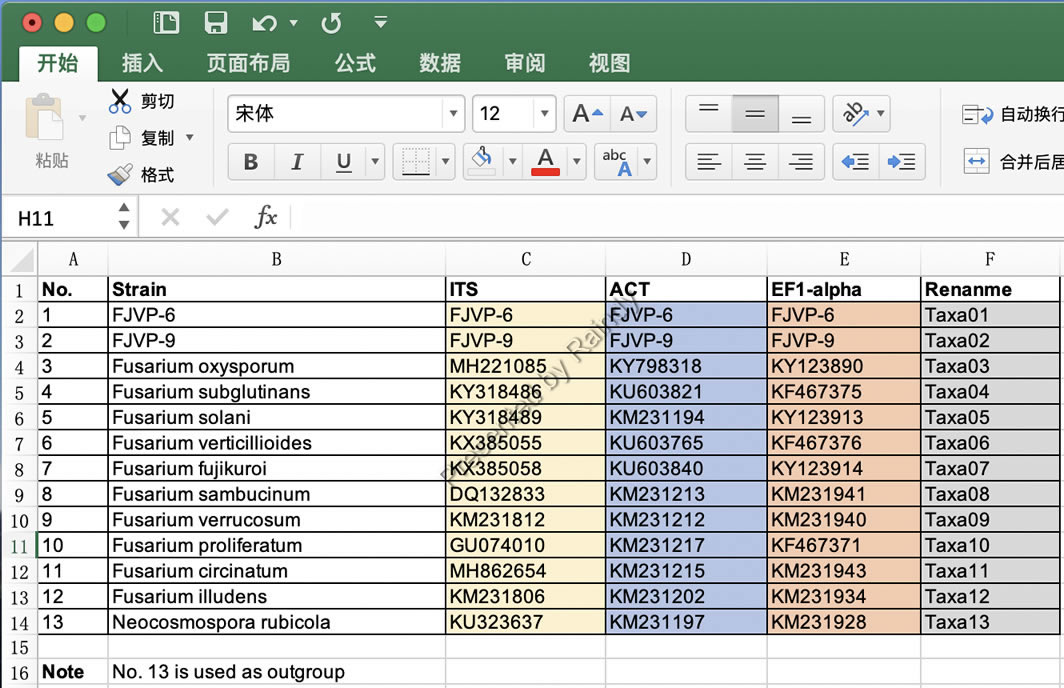
(2)整理好的Excel文件,参考格式如下图所示,第一列为序号,第二列为菌株名称(长文件名称),第三列-第五列为不同基因数据,最后一列为重命名的名称
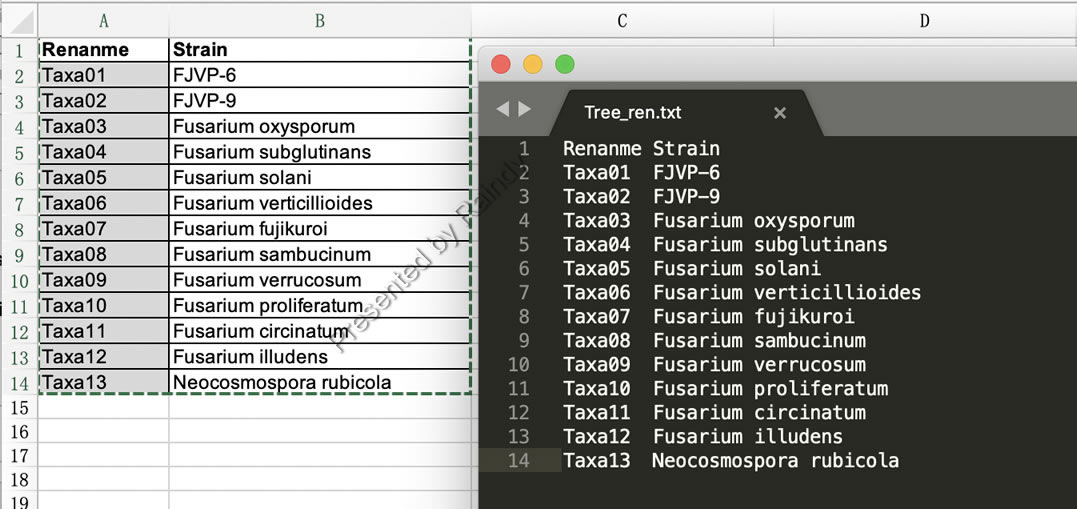
Raindy注:第二列长名称是用批量重命名树名称用。此外,多基因联合时需要名称一样才可以串联,所以最后一列需要统一命名为相同名称。
简明流程:
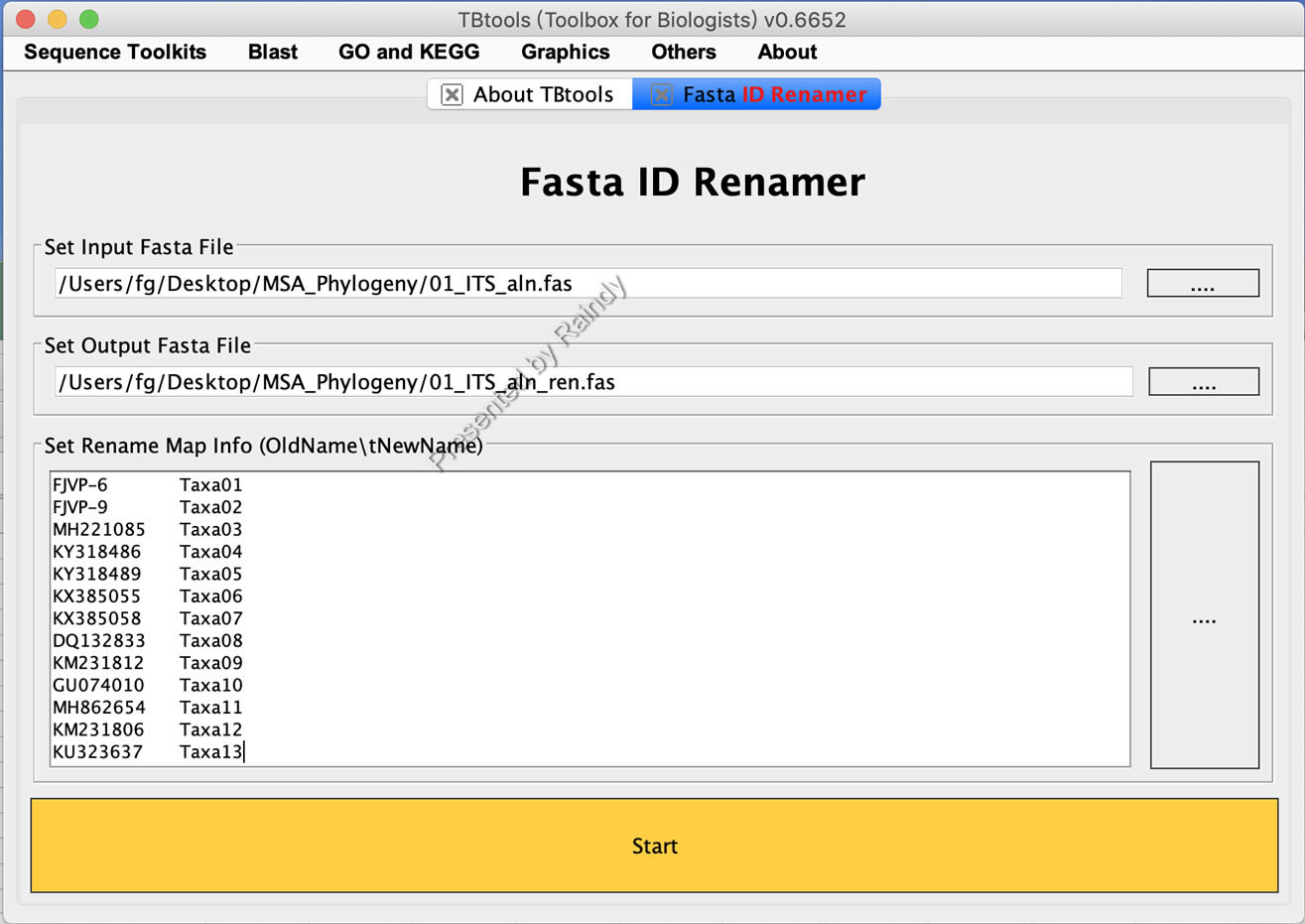
Step 1: 将不同基因中的序列名称批量为统一名称,可以使用TBtools中的Fasta ID Renamer进行,如下图所示,将待比对后待重命名的序列通过拖曳到Input Fasta File后的文本框,设置输出FASTA文件名称(示例为:01_ITS_aln_ren.fas)后,将重命名名称对照表粘贴入Rename Map Info后的文本框。
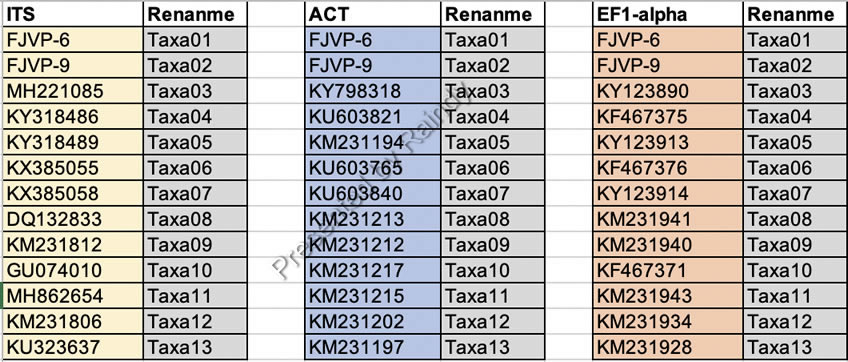
重命名的对照表信息可以从事前面的Excel文件中整理而来,如下图所示:
Step 2:串联不同基因序列文件,运行PhyloSuite后,在Alignment菜单下,点击“Concatenate Sequence”弹出操作界面,将待串联的不同基因序列拖入“Input”后的文本框内,如需调整前后顺序,可以选择对应基因序列文件后按住不放,拖到正确位置即可。
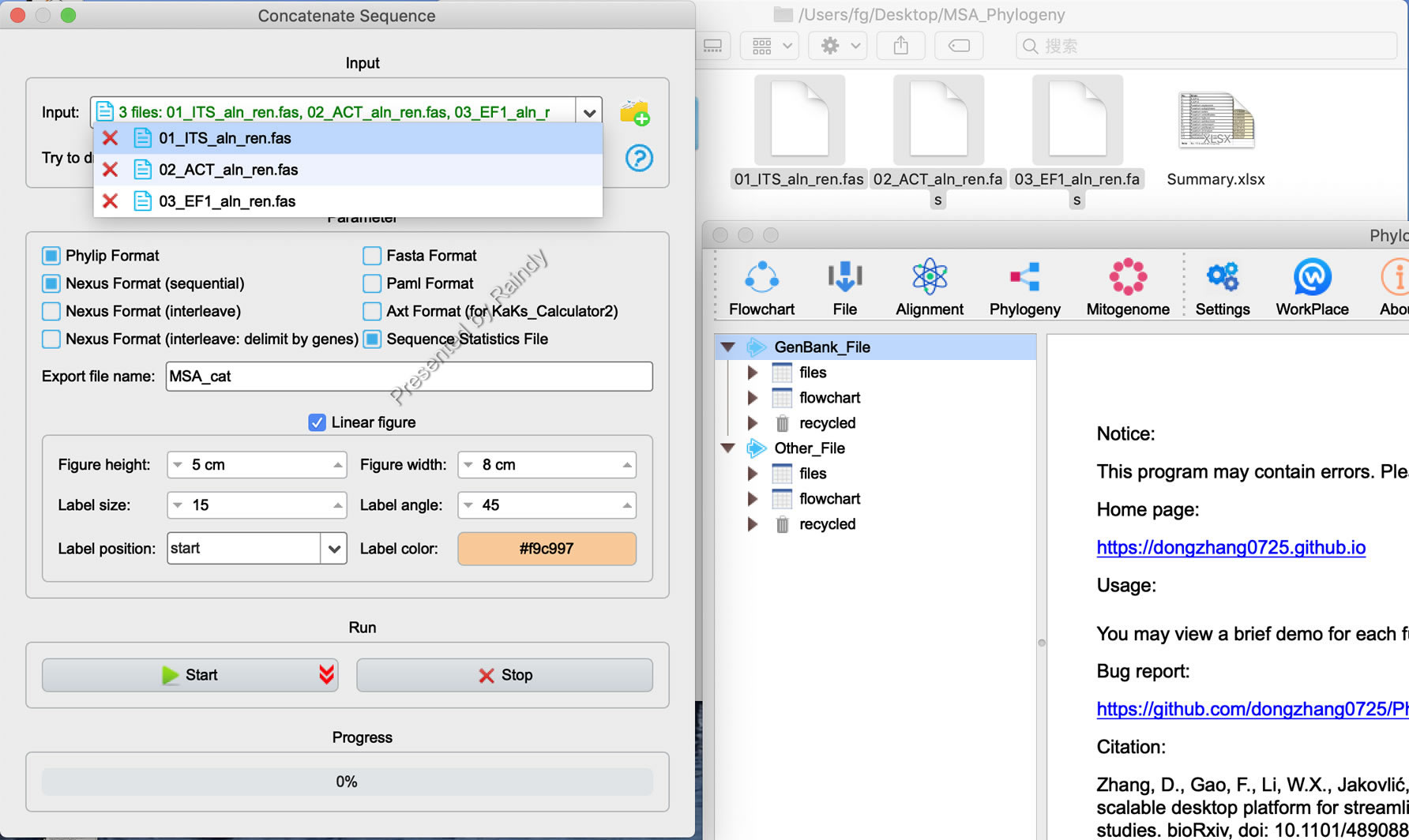
点击“Start”后,程序自动开始串联,如果不同基因数据缺失,会弹出相应提示,“Show Details”可以查看具体信息并进行检查。
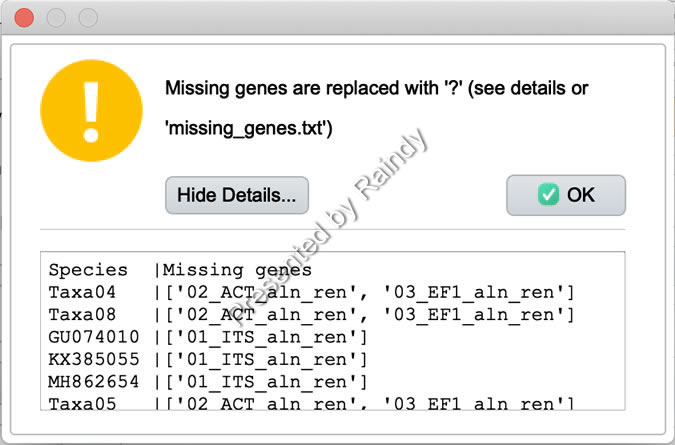
如果不同基因序列信息完整,合并完成弹出信息如下图所示。
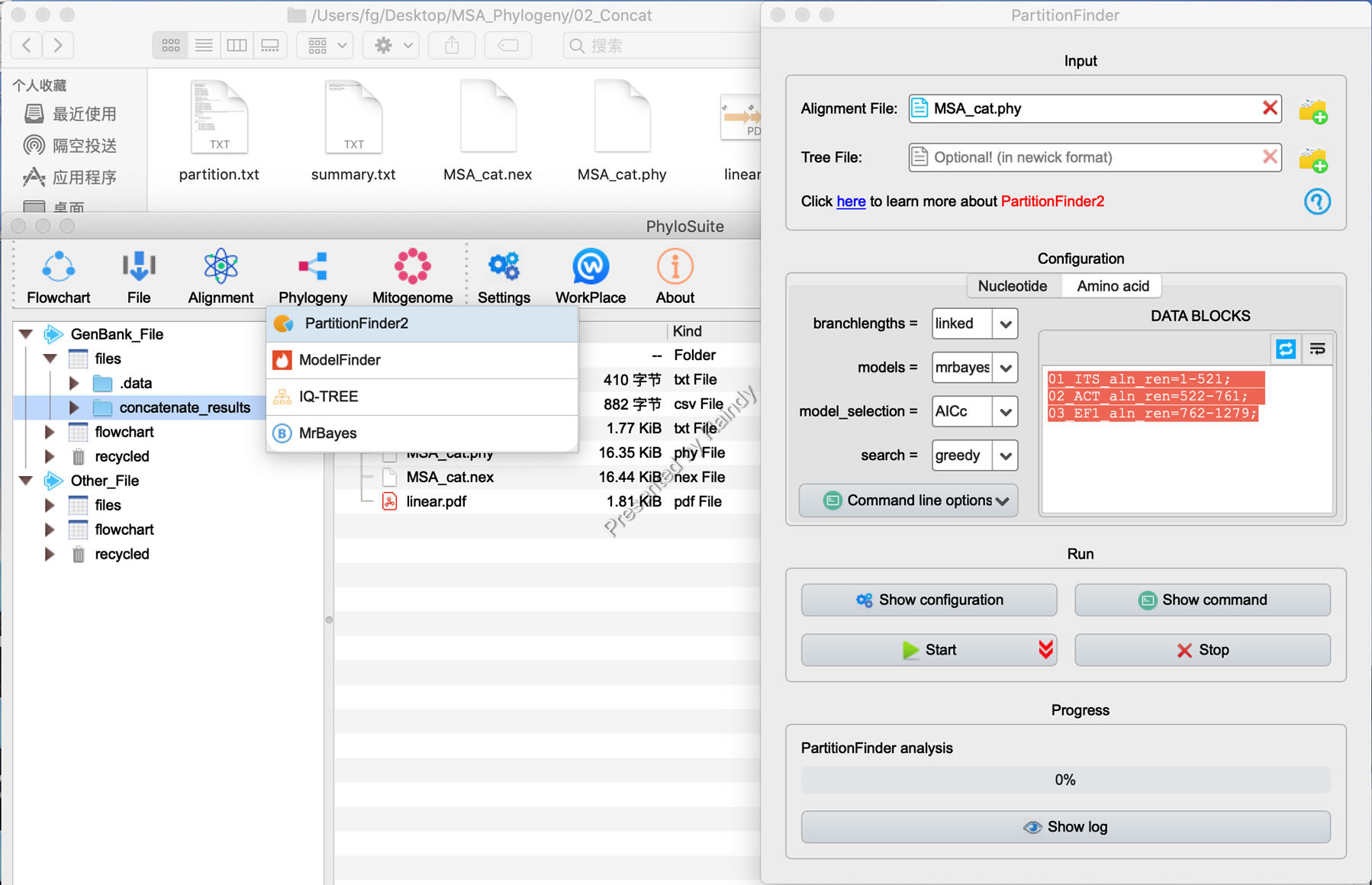
Step 3: 确定多基因数据分区的最适模型方案,依次点击PhyloSuite的“Phylogeny”菜单下的“PartitionFinder”,弹出界面如下图,前一步的合并会自动被读入到Alignment File后的文本框内,注意branchlengths=linked(大部分情形如是),Models后选择“MrBayes”,Model_selection选择AICc(PartitionFinder作者推荐),Data Blocks 也会自动识别为不同基因分区。如果需要进一步Codon分区,可以选择对应基因后,点击DATA Blocks下方的蓝色双向箭头即可。示例数据只按基因分区,不对ACT和EF1基因进一步Codon分区。
Step 4: 多基因联合重建贝叶斯树,依次点击“Phylogeny”菜单中的“MrBayes”,同样方式,前一步数据和分区模型参数会自动被读取到弹出的MrBayes参数设置界面,Outgroup通过下拉菜单选择Taxa13,其他参数可以使用默认值,当然可以根据实际调整。
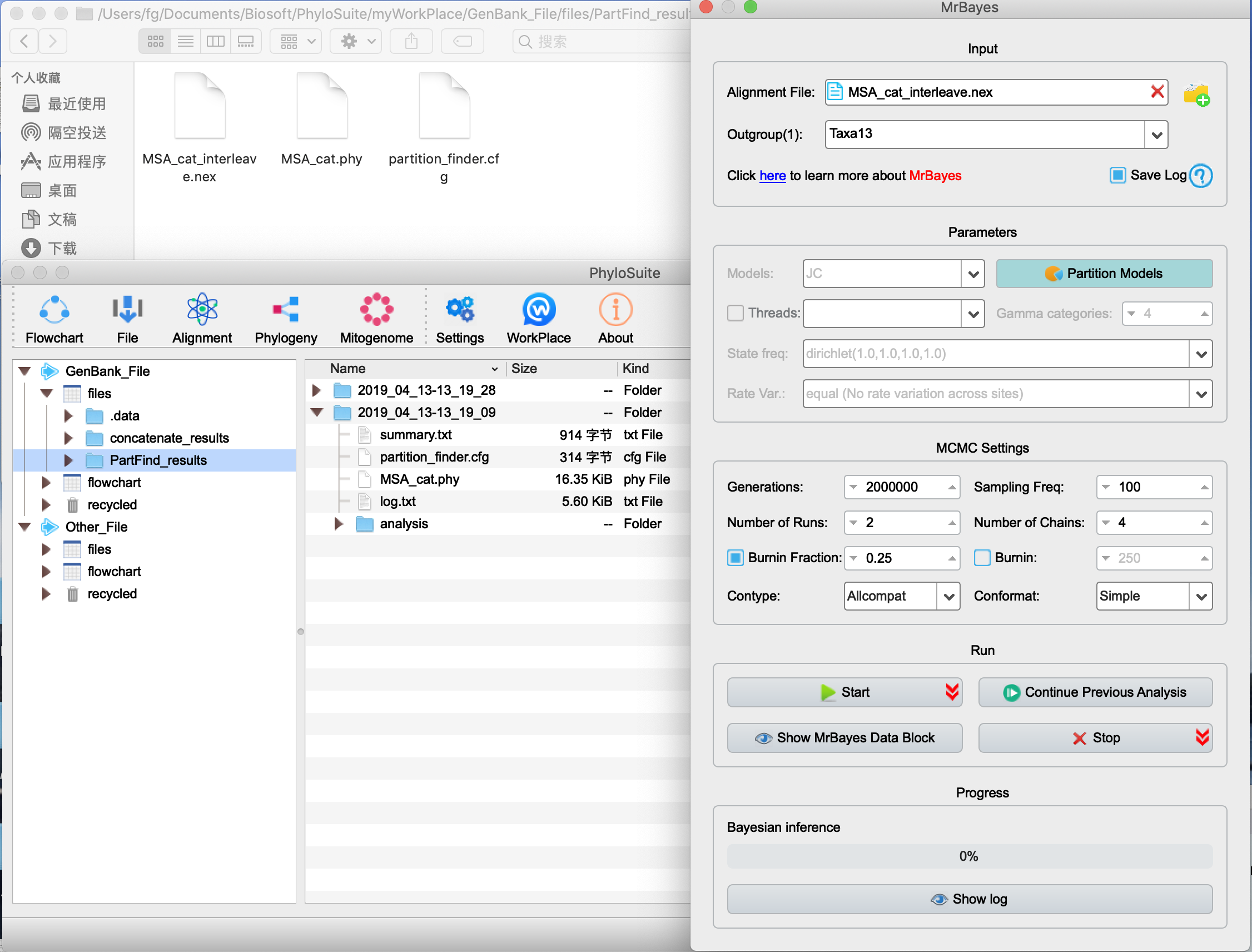
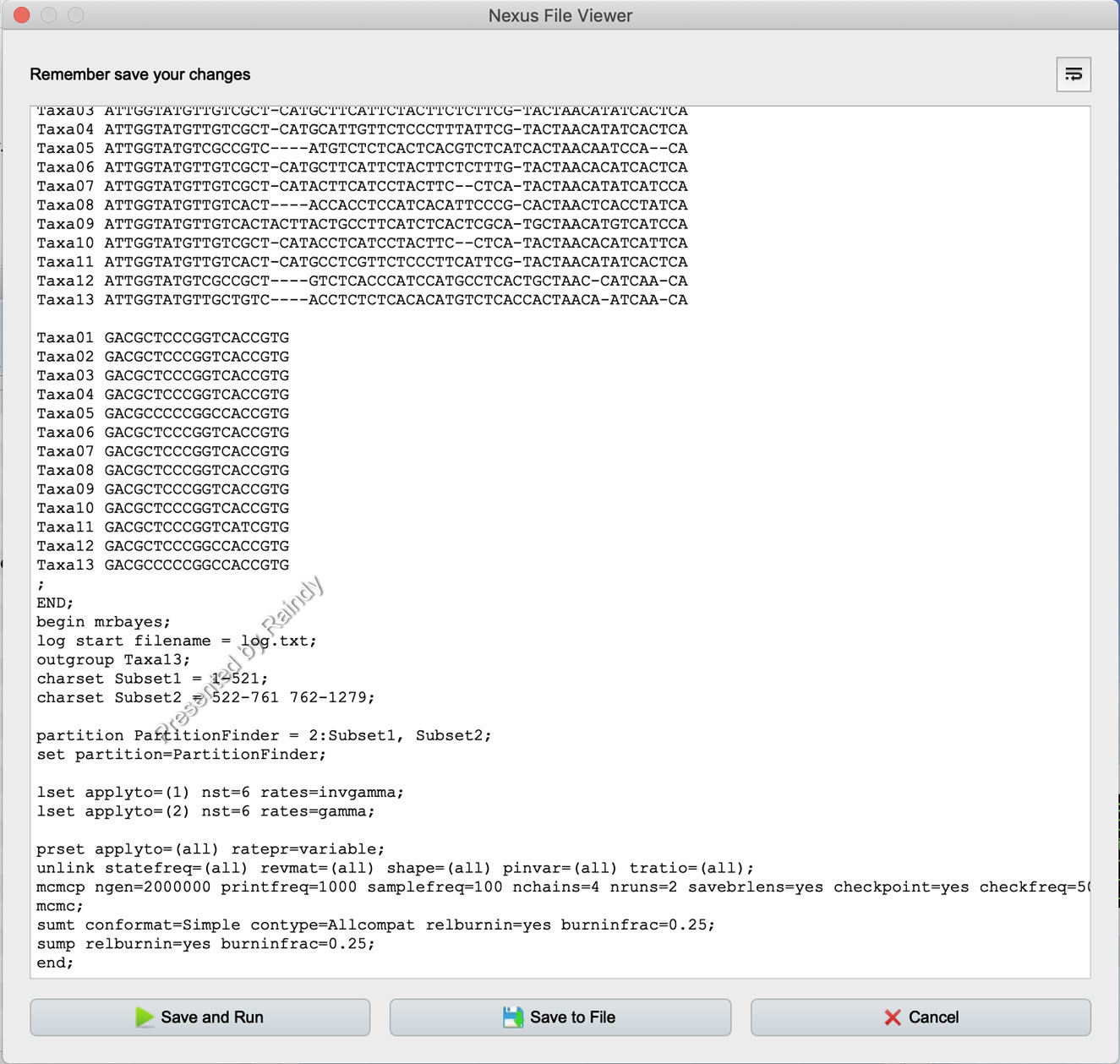
当然,PhyloSuite中的MrBayes参数设置界面提供一下Nexus File Viewer查看器,可以随时查看相关运行参数和模型信息等。
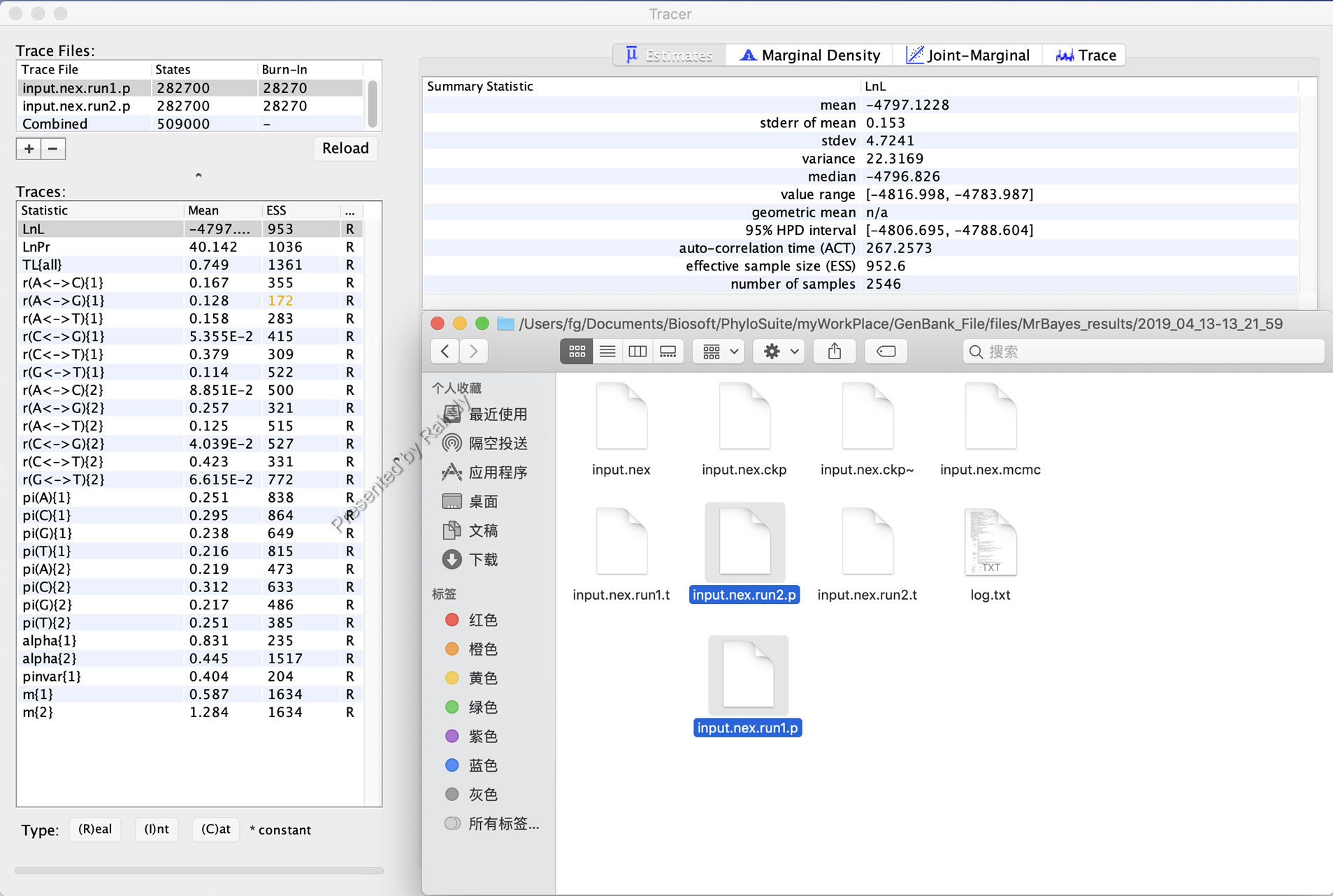
MrBayes分析完成后,可以通过两种方法查看参考是否收敛,一是用Tracer软件查看xxx.runx.p文件,如下图所示:
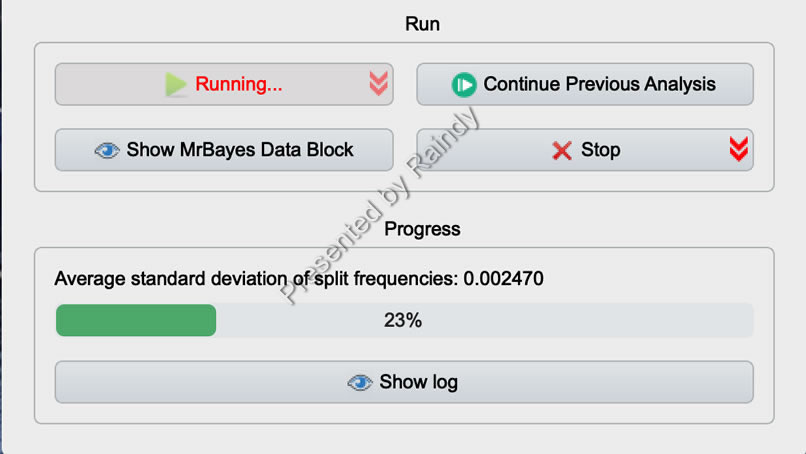
二是直接查看Average standard deviation of split freq. 是否显著低于0.01(如果达不到,0.05也可以接受)
Step 5: 查看树文件并批量恢复树长名称,在FigTree查看input.nex.con.tre前,准备一下树名称的重命名对照表(也可以从Excel文件中整理出来),并保存为Tree_ren.txt 如下图所示:
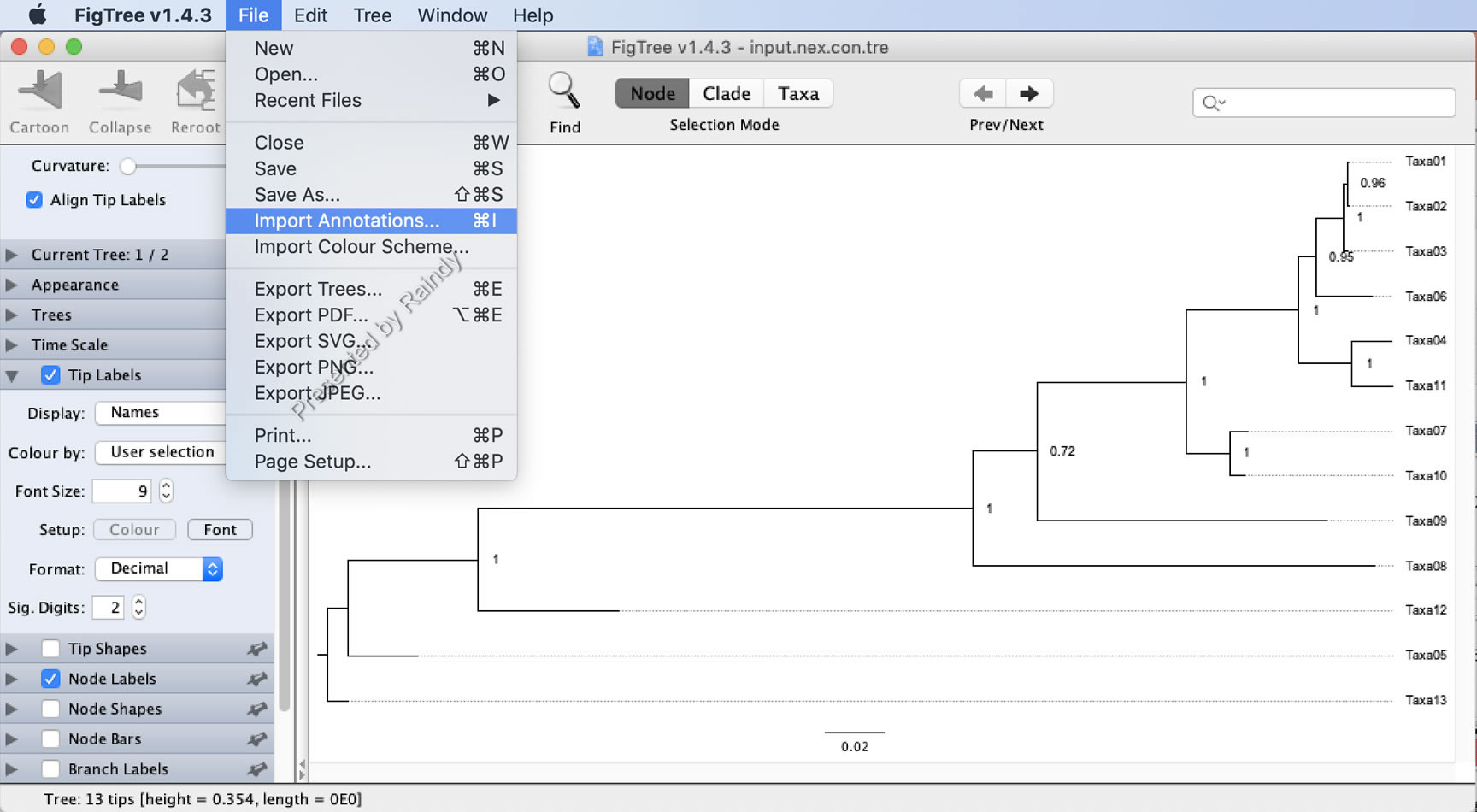
在FigTree中载入贝叶斯树文件后,基本设置完毕(如:Node Label 等),在“File”菜单“Import Annotations”选择前面准备的tree_ren.txt文件,并在“Tip Labels”的“Displaye”下拉菜单选择“Strain”(名称示实际情况不同,示例数据为Strain)。
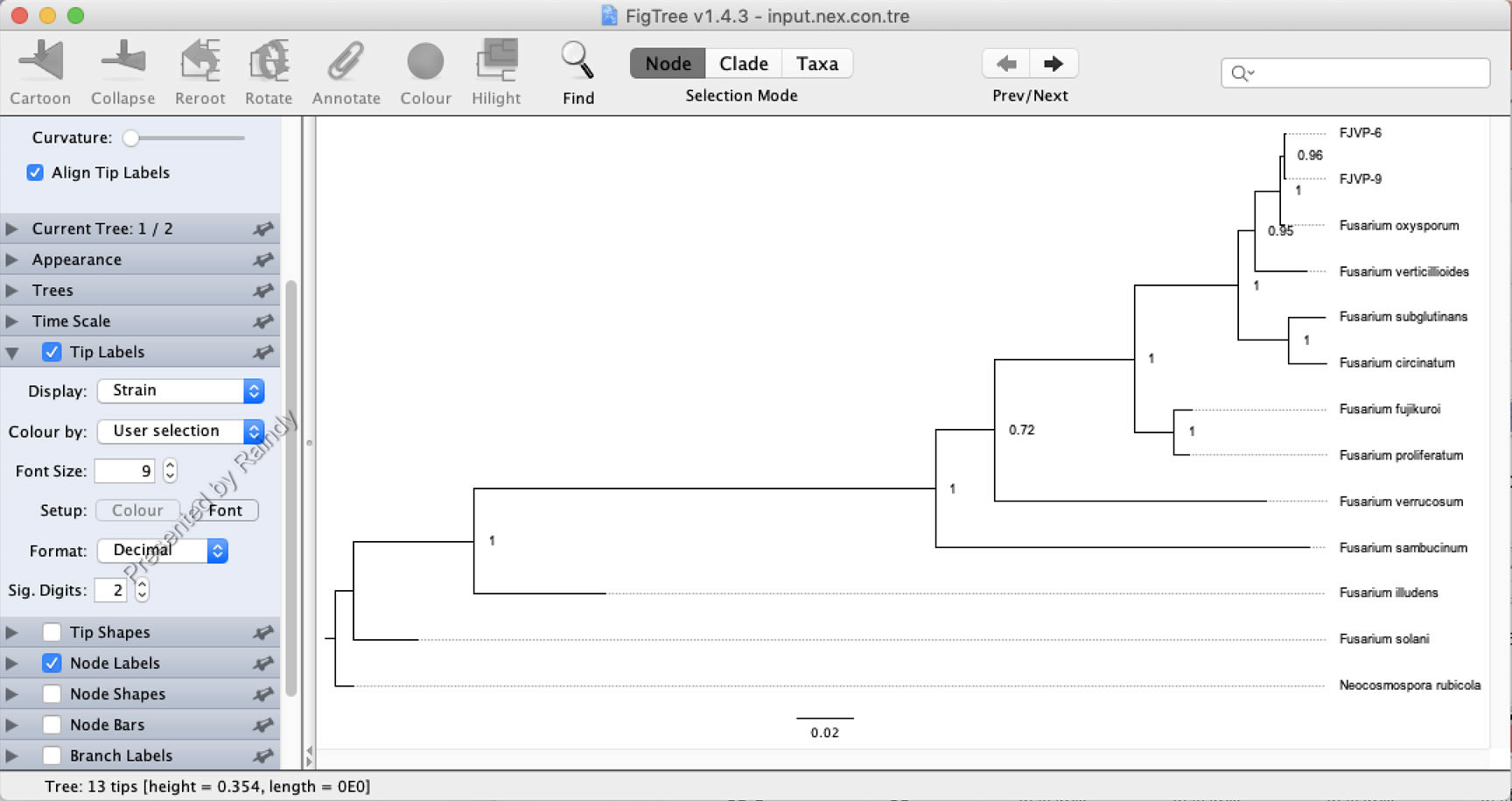
切换“Strain”后,树的Tip名称批量显示为长名称,比较直观,如下图所示
注意事项:
重命名序列时,需要注意检查GenBank登录号后是否存在空格,如有请替换为空,否则批量重命名不成功

链接:
多基因联合分析简明图解之IQ-TREE篇:
https://user.qzone.qq.com/58001704/blog/1517027454
多基因联合建树图解教程(更新):
https://user.qzone.qq.com/58001704/blog/1396317591
PhyloSuite相关资源:
https://dongzhang0725.github.io/
https://blog.sciencenet.cn/blog-460481-1173109.html
上一篇:应用贝叶斯谱系动力学解析我国TMV的时空迁移特征
下一篇:PopART 绘制 Haplotype Network 图解(By Raindy)