博文
低覆盖度基因组测序获取系统发育组学数据
 精选
精选
||
低覆盖度基因组测序获取系统发育组学数据
Phylogenomics from Low-coverage Whole-genome Sequencing
Zhang, Feng; Nanjing Agricultural University, Department of Entomology, College of Plant Protection
Ding, Yinhuan; Nanjing Agricultural University
ZHU, Chao-Dong; Institute of Zoology, Chinese Academy of Sciences
Zhou, Xin; Beijing Advanced Innovation Center for Food Nutrition and Human Health, China Agricultural University; Department of Entomology, China Agricultural University
Orr, Michael; Key Laboratory of the Zoological Systematics and Evolution, Institute of Zoology, Chinese Academy of Sciences
Scheu, Stefan; Georg-August-Universitat Gottingen, J.-F.- Blumenbach Institute of Zoology & Anthropology
Luan, Yunxia; School of Life Sciences, South China Normal University
Methods in Ecology and Evolution(accepted on 2018/12/21)
方法细节:
论文链接:
2019-Phylogenomics from low-coverage whole-genome sequencing.pdf
英文摘要:
1. Phylogenetic studies are increasingly reliant on next-generation sequencing (NGS). Transcriptomic and hybrid-enrichment sequencing techniques remain the most prevalent methods for phylogenomic data collection due to their relatively low demands for computing powers and sequencing prices, compared to whole genome shotgun sequencing (WGS). However, the transcriptome-based method is constrained by the availability of fresh materials and hybrid enrichment is limited by genomic resources necessary in probe designs, especially for non-model organisms.
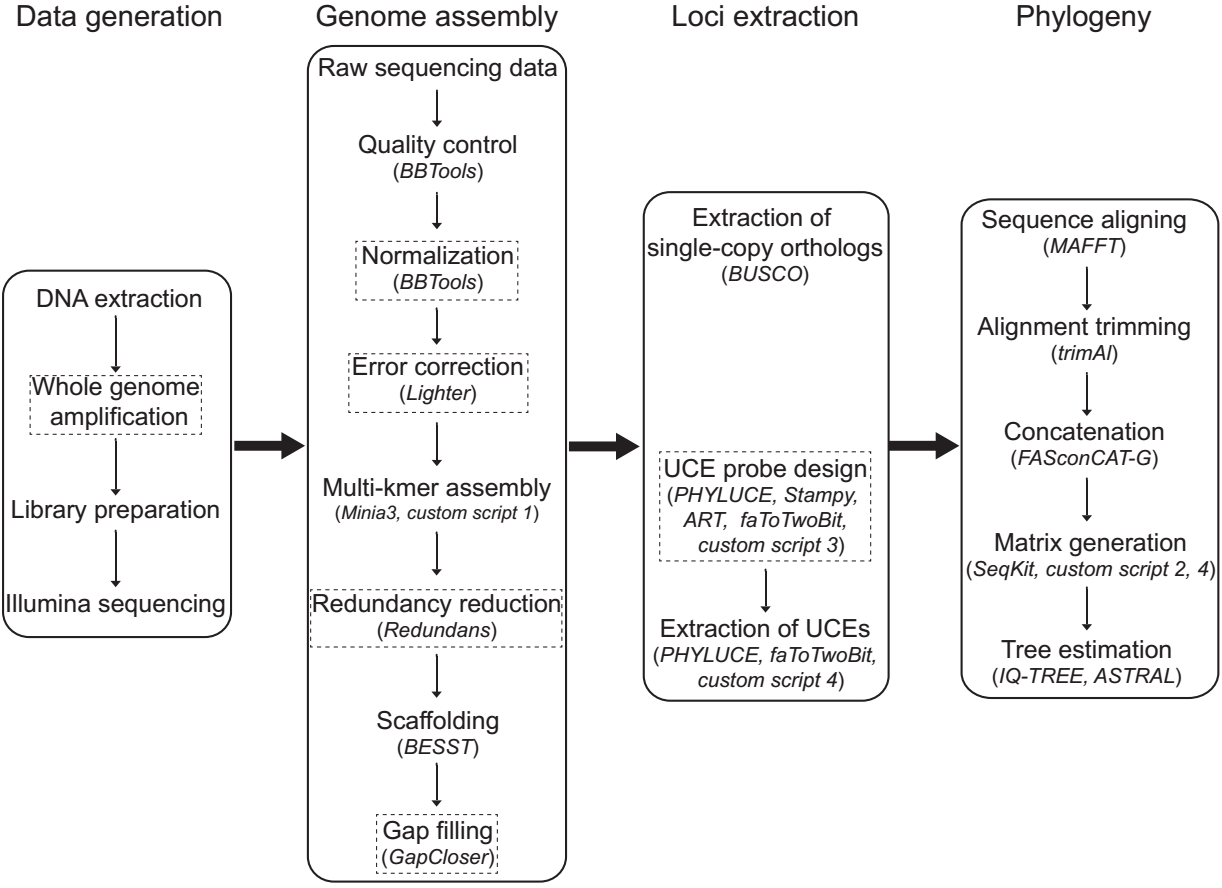
2. We present a novel WGS-based pipeline for extracting essential phylogenomic markers through rapid de novo genome assembling from low-coverage genome data, employing a series of computationally efficient bioinformatic tools. We tested the pipeline on a Hexapoda dataset and a more focused Phthiraptera dataset (genome sizes 0.1‒2 Gbp), and further investigated the effects of sequencing depth on target assembly success rate based on raw data of six insect genomes (0.1‒1 Gbp).
3. Each genome assembly was completed in 2‒24 hours on desktop PCs. We extracted 872‒1,615 near-universal single-copy orthologs (BUSCOs) per species. This method also enables development of ultraconserved element (UCE) probe sets; we generated probes for Phthiraptera based on our WGS assemblies, containing 55,030 baits targeting 2,832 loci, from which we extracted 2,125‒2,272 UCEs. Resulting phylogenetic trees all agreed with currently-accepted topologies, indicating that markers produced in our methods were valid for phylogenomic studies. We also showed that 10‒20× sequencing coverage was sufficient to produce hundreds to thousands of targeted loci from BUSCO sets, and even lower coverage (5×) was required for UCEs.
4. Our study demonstrates the feasibility of conducting phylogenomics from low-coverage WGS for a wide range of organisms without reference genomes. This new approach has major advantages in data collection, particularly in reducing sequencing cost and computing consumption, while expanding loci choices.
中文摘要:
1. 系统发育研究越来越依赖于新一代测序(NGS)技术。与全基因组鸟枪测序法(WGS)相比,转录组学和杂交富集测序技术是目前用于系统发育基因组数据收集的最常用方法,对计算能力和测序价格的需求相对较低。然而,转录组测序需要新鲜材料,杂交富集测序需要类群特异的探针设计,这些限制在非模式生物尤其明显。
2. 我们提出了一种新的基于WGS的分析流程,可以从低覆盖度测序[MS2] 的基因组数据中快速从头组装基因组,并使用一系列计算效率高的生物信息学工具来提取常用的系统发育基因组标记。我们在六足动物(Hexapoda)和虱目(Phthiraptera)数据集(基因组大小0.1-2 Gbp)中测试了流程,并基于6个昆虫基因组(0.1-1 Gbp)进一步研究了测序覆盖度对分子标记组装成功率的影响。
3. 每个基因组装配在台式PC上仅用时2-24小时。每个物种提取了872-1,615个通用单拷贝直系同源基因(BUSCO)。该方法还能够开发超保守元件(UCE)探针:我们基于WGS组装结果设计了虱目探针,包含针对2,832个UCE标记的5530个诱饵,并以此提取了2,125-2,272个UCE。基于这些数据重建的系统发育树都与目前公认的拓扑结构一致,表明该方法产生的标记对系统发育学研究是有效的。我们还发现,10-20倍的测序覆盖度足以产生数百到数千个靶向BUSCO基因,而UCE可能需要更低的覆盖度(5×)。
4. 我们的研究证明了在没有参考基因组的情况下,从低覆盖度WGS测序开展系统发育基因组学的可行性。这种新方法在数据收集方面具有显著优势,特别是在降低测序成本和计算消耗的同时,提升了分子标记选择的范围。
致谢:
本项工作得到国家自然科学基金委面上项目(31772491、 31772510)、中国科学院动物进化与系统学重点实验室资助(Y229YX5105)。朱朝东及其实验室得到国家自然科学基金委杰出人才项目支持(31625024)。
https://blog.sciencenet.cn/blog-536560-1153280.html
上一篇:FitCons:进化论指导表观遗传组数据分析
下一篇:Journal Club:性别决定、性别比例和遗传冲突(John H. Werren,1998)