博文
[转载]胎儿染色体非整倍体无创DNA检测原理
|| |
文章转自:http://blog.sciencenet.cn/blog-306699-944921.html
目前基于新一代测序技术(Next Generation Sequencing, NGS)在临床充分利用的产前筛查方法(或者叫唐氏筛查)的原理最早在PNAS的两篇文章,通在2008年上发表,相关的数据处理策略略有差别,两篇文章分别是“Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma” 和“Noninvasive diagnosis of fetal aneuploidy by shotgunsequencing DNA from maternal blood“,两个团队基于各自的技术手段,先后都成立了提供唐氏筛查的技术服务公司。
文章"Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma"中的数据处理原理如下:
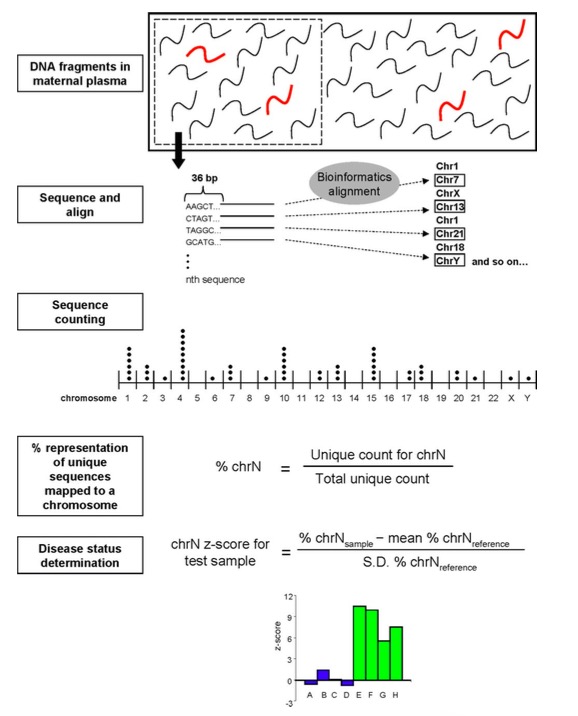
第一步:Fetal DNA (thick red fragments) circulates inmaternal plasma as a minor population among a high background of maternal DNA (black fragments). A sample containing a representative profile of DNA molecules in maternal plasma is obtained. 在母体血液中有游离的胎儿DNA片段,在上述途中,红色的片段代表胎儿DNA片段。从图上可以看出,母体血液中胎儿DNA的浓度非常低,为此科研工作者的目的在于用数据处理的方法能够把信息放大到准确检测的地步。
第二步: In this study, one end of eachplasma DNA molecule was sequenced for 36 bp using the Solexa sequencing-by-synthesis approach. 当时用Solexa测序技术测取36bp的reads,目前主要基于Illumina、CG和Proton的测序技术,读长和深度都有所增加。
第三步:The chromosomal origin of each 36-bp sequence was identified through mapping to the human reference genome by bioinformatics analysis.将测取到的reads进行mapping操作,比对到人的参考序列上。
第四步:The number of unique sequences mapped to each chromosome was counted and then expressed as a percentage of all unique sequences generated for the sample, termed % chrN for chromosome N. 统计每条染色体上uniq比对的reads数目,同时计算每条染色体上uniq reads的比例。
第五步:Z-scores for each chromosome and each test sample were calculated using the formula shown. 计算一个z-score值,利用染色体上uniq reads的比例减去control样本中对应此条染色体uniq reads比例的平均值,再除以control样本中此条染色体uniq reads的标准差。例如,21三体筛查, 假设有100个已知怀正常胎儿的样本,通过前4步的计算,可以得到每个样本在21号染色体上,uniq reads的比例具体值,如此可以计算这100个样本在21号染色体的平均值和标准差。如果1个新的样本要判断是不是21三体,可以根据上述公式去计算21号染色体的z-score,超过界定界限则是患病胎儿,反之正常。其中就可以看到,正常样本数越多,平均值和标准差就越准(基线越来越标准),如此得到的z-score更准确。
通过以上方法最终的结果如下:

从上图可以看出,如果利用染色体上的uniq reads比例不能很好区分正常胎儿和3体胎儿,利用z-score的计算,可以发现3体胎儿的z-score超过3,正常胎儿都位于3以上,为此达到很理想的检测目的。
文章"Noninvasive diagnosis of fetal aneuploidy by shotgunsequencing DNA from maternal blood"中的数据处理原理和结果如下:
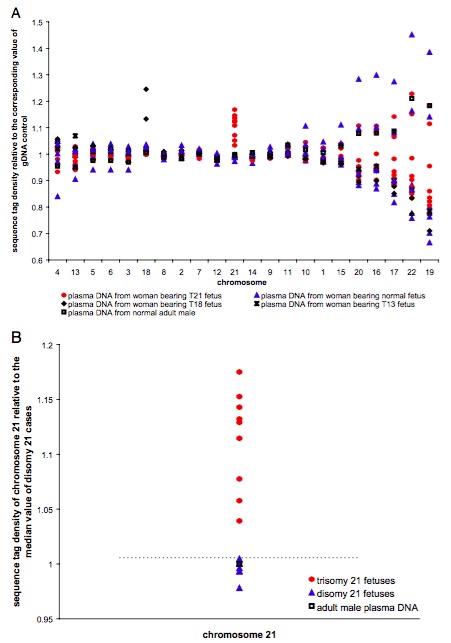
第一步:We obtained on average 10 million 25-bp sequence tags per sample. 测序获取数据;
第二步:将每条染色体按50kb划分bin,根据bin中uniq reads的数目进行排序,取中位数代表此条染色体的reads数目;
第三步:每个样本有21条染色体的reads数目,再对21个数据排序,取中位数代表此样本的reads数目;
第四步:用每条染色体的reads数目除以每个样本的reads数目得到归一化的一个值;
第五部:利用第四步得到的诡异化值去计算置信区间,当样本的值不在此置信区间时,被判定为异常样本。
利用此处理策略,可发现针对21三体,能够较理想区分正常胎儿和异常胎儿样本。此处理策略也依赖于正常样本的数目,正常样本越多,计算得到的置信区间会越准确,判断的结果也就会越准。
参考文章:http://www.pnas.org/content/early/2008/10/03/0808319105
http://www.pnas.org/content/105/51/20458.abstract
https://blog.sciencenet.cn/blog-682704-1171063.html
下一篇:[转载]新生儿遗传病基因筛查技术及相关疾病简介