博文
不知道miRNA测序数据接头就没法继续分析了吗?教你利用clustalw和blat解决接头问题
|
miRNA是一类长约22 nt的非编码小RNA。miRNA通过碱基互补配对结合到靶mRNAs 3’UTRs以调控数百个基因的表达。因此,miRNA参与几乎所有生命活动,与生物体的生长、发育、疾病、衰老和死亡息息相关,可以作为疾病诊断和检测的生物标志物。
日常数据分析中,我们经常会遇到各种来源的miRNA测序原始数据,有些测36 bp,有些测50 bp,有些测75 bp或者更长的。形形色色的reads长度,adaptor接头序列,众多测序公司来源的数据,给数据分析带来了诸多不便。其中比较常见的问题就是:接头序列是什么?
小编就曾经遇到过这个问题,问某公司:“你们测序的接头序列是什么”?答复:“保密”!小编当时就想怼他:不知道接头序列,就没法把接头去掉,不去掉接头怎么往下分析?都是一个圈子混的,保不保密我不知道吗?
今天小编就教你如何在不知道接头序列的情况下,把接头序列从原始fastq文件中扒出来,同时把read的组成结构也分析一下。
1,fastq数据格式
 图1. fastq格式
图1. fastq格式
Fastq格式是测序原始数据存储标准格式,存储了每条read的序列及碱基质量分数。如图1所示,每4行代表一条read。
以Read1为例:
第1行代表序列的编号,以@开头;一般包括:机器编码(A00212),run id(123),flowcell编号(H3TTTDSX3),lane号(4),flowcell上的tile编号(1101),X坐标(8919)和Y坐标(1000),其后是barcode信息,用于将多个样品拆开(demultiplex)。
第2行是read序列,一般ATCG,有时候有N。N表示荧光信号干扰无法判断到底是什么碱基。
第3行是识别码,以+开头;后边接序列标示符、描述信息等,或者干脆就是+
第4行是碱基质量分数,与read序列一一对应,即每个碱基有一个分数,表明该碱基的可信程度。由于不同型号机器出来的值不太一样,因此可以根据这些值大概判断测序仪型号。
2,miRNA read构成
通常,miRNA read有三种构成方式(如图2所示):
A:miRNA序列后边直接连接adaptor接头序列
B:miRNA序列前面加几个碱基,后边接miRNA序列,然后连接adaptor接头序列
C:miRNA序列后边加几个碱基,然后连接adaptor接头序列
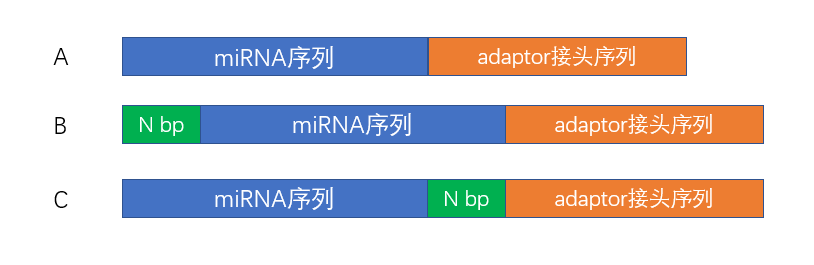
图2. miRNA read构成
3,miRNA常见接头序列
通常illumina测序的小RNA接头序列有:TGGAATTCTCGGGTGCCAAGGAACTCC、AACTGTAGGCACCATCAAT、AGATCGGAAGAGCACACGTCT等,拿到原始数据后,首先可以先用这3种序列去搜下。搜到了就可以先用这些接头去预分析下,拿到去接头后的miRNA长度分布图,如果长度分布正常(在22nt)附近有峰,则表示接头序列正常去掉,这种一般是图2中的A情况。对于B和C就无能为力了。
4,Clustalw检测接头序列
4.1 fastq collapse
在完全未知的情况下,我们可以这样操作。首先,将fastq格式转成fa格式,并且对每条不同的read序列进行计数,所谓的collapse步骤。
可以使用Galaxy在线工具进行操作。https://usegalaxy.org/,搜索collapse,上传fa或者fastq文件即可,其功能是统计每条unique序列的个数
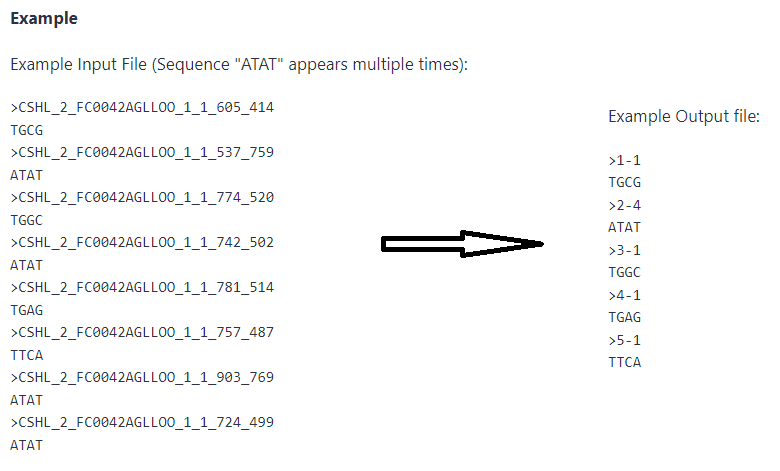
图3. read序列collapse
以小编手里的fastq文件为例,经过collapse后,前10条序列为:

图4.top 10 read
肉眼可见,中间的部分序列在每条read中都存在,可能是潜在的接头序列。
4.2 clustalw多重序列比对
使用在线clustalw多重序列比对软件来检查下我们的top10序列。一般情况下,如果实验做的没问题,个别miRNA的表达量会很高,它们可能占据了绝大部分的reads,因此我们可以利用这个大概率来进行校验。
图5. Clustalw输入top10序列
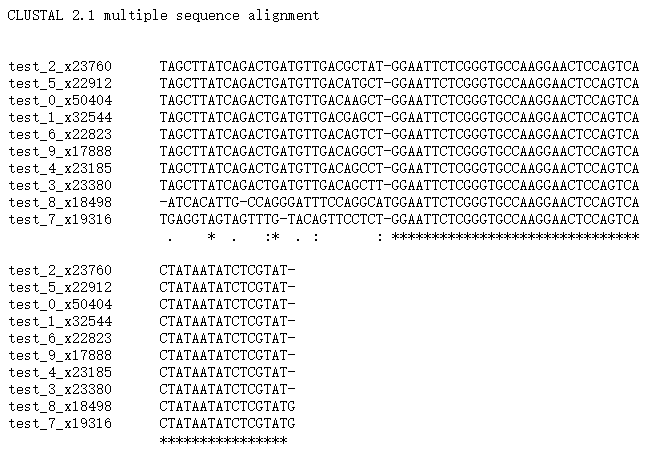
图6. 多重序列比对结果
其中星号为所有序列中都一样的碱基。可以看出,后边的星号连续排列,因此初步判断为接头序列,而星号前面一个碱基仅在test_8里边与其他的9条不一样,因此,我们也把T作为接头的一部分,于是预测的接头序列就是TGGAATTCTCGGGTGCCAAGGAACTCC……,经过比较,发现与我们上面提到的接头序列一样,因此,可以完全确定接头序列是正确的。
5,blat比对验证read组成
在找到接头序列后,我们可以将接头序列及以后的碱基全去掉,这样我们就得到了可能包含miRNA的reads。

图7. 包含接头的reads
如图7所示,绿色为接头序列,其左侧为可能的miRNA序列,将这10条去接头的序列(图8),粘贴到UCSC genome browser的blat工具中,检索这些序列。

图8. 去掉接头的序列
图9. Blat提交页面
Blat结果如下:
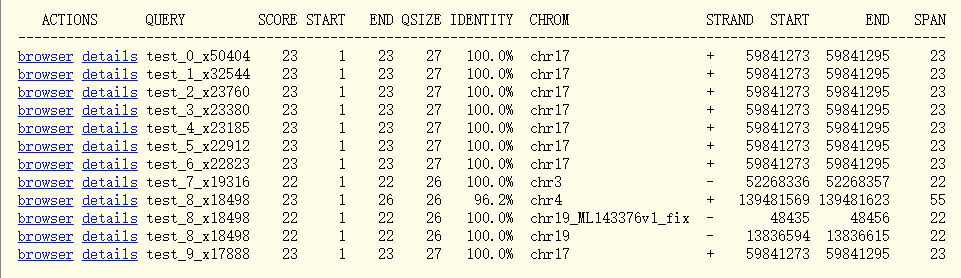
图10. Blat结果
从图10的blat结果可以看出,我们查询的序列是26或者27 bp,其中identity为100%的序列长度跨了22或者23 bp,因此可以推测序列前或者后加了4 bp的短序列。
打开details链接(图11)查看详细比对结果,可以看出都是后边4 bp的短序列没有比对上。

图11. blat详细比对情况
6,结论
因此,综合上面clustalw鉴定的接头序列,和blat鉴定的read组成,我们得出接过来:该fastq的read组成是图12的形式。Read结构分析清楚后,我们就可以按部就班地进行后续的表达、差异、靶基因等分析了。

图12. Read构成
总结:认清问题,寻找解决问题的思路,思路清晰后,就可以找软件工具进行处理。
微生信助力发文章,谷歌引用630+,知网引用470+
https://blog.sciencenet.cn/blog-707141-1355647.html
上一篇:来了,来了!GSEA官网更新,Mouse基因集终于有了“官方认证”
下一篇:ChIP-Seq,MeRIP-seq峰(peak),eccDNA等基因组位置染色体分布可视化