博文
MarkerMap:单细胞研究的非线性标志物选择
||
MarkerMap:单细胞研究的非线性标志物选择
基因组学和显微镜技术的最新进展使得从空间和时间坐标收集细胞间的单细胞基因表达数据(scRNA-seq)成为可能。了解细胞如何跨越时空尺度聚集信息,以及反过来,基因表达变异性如何反映这种聚集过程仍然具有挑战性。一个特殊的实验设计挑战是由于现有技术(例如smFish, seqFish, MERFISH, ISS)依赖于少量靶基因或标志物的预先选择,无法捕获表征细胞群体细微差异所需的完整转录组学信息。选择最好的标志物(标志物选择)通常在统计和计算上具有挑战性,通常是数据非线性和要捕获的差异类型的函数。
标志物选择是先验知识和对先前收集的scRNA-seq数据进行计算分析的产物。在计算上,它旨在降低数据的维度,如基因表达——从数千个基因到几个——以实现下游分析,如可视化、细胞类型恢复、基因程序鉴定或介入研究的基因面板设计。类似于主成分分析(PCA)或变分自编码器(VAE),这两种方法在单细胞RNA分析中都很流行,标志物选择方法试图将细胞描述为少数坐标空间中的数据点。为此,基于PCA和VAE的方法将细胞与一组较小的潜在坐标关联起来,这些潜在坐标表示基因表达的加权组的聚集。相比之下,标志物选择方法寻求可解释的表示,其中坐标直接表示基因,而不是线性或非线性的基因组合。
已经提出了许多方法来选择标志物,以最好地区分一组离散的,预定义的细胞类型类别。这些方法可分为两大类:一对全和基因面板方法。“一对全”方法是最常见的,这种方法旨在确定每种细胞类型的一组基因,这些基因仅在这种细胞类型中与所有其他细胞类型相比有差异表达。特别是RankCorr,一种受相关蛋白质组学应用成功启发的稀疏选择方法,提供了理论保证和出色的实验性能。另一个最近表现良好的算法SMaSH使用了神经网络框架,利用了可解释的机器学习文献中的技术。相比之下,基因面板方法寻求识别共同区分细胞类型的遗传标志物组。 例如,ScGeneFit是一种压缩分类方法,它采用线性规划来选择保留数据分类结构的标志物,而不需要识别具有单个细胞类型的基因,因此可能选择更少的基因。这些方法都是有监督的:它们依赖于细胞的真实分类结构。SCMER是唯一一种通过非线性降维(UMAP)和流形学习来避免显式聚类的遗传标志物选择方法。 最近关于基因组学应用中的特征选择的综述比较和对比了这些标志物选择方法在有监督的线性环境下的应用。
更广泛地说,已经提出了不同的解决方案来解决非基因组背景下的特征选择问题。在线性设置中,这些包括流行的正则化或Lasso和CUR分解,而在非线性回归设置中,结果通常使用神经网络预测。在语言模型中,可解释的深度学习算法已经被开发出来,用于预测和解释结果,如评论评级或采访结果,这些结果来自文本,其中很少有重要的单词被突出显示为结果的解释。在成像中,给定一个训练好的模型,可以使用shapley系数来识别图像中产生某种预测的部分。
最近,Gregory等人介绍了MarkerMap(图1,https://github.com/Computational-Morphogenomics-Group/MarkerMap),一个可扩展和生成的非线性标志物选择框架。MarkerMap的目标是双重的:(a)提供一种通用的方法,允许联合标志物选择和全转录组重建,以及(b)在一个单一的,可访问的计算框架内,围绕转录组研究,比较和对比不同社区的工具(包括计算生物学和可解释的机器学习)。因此,MarkerMap展示了几个关键特性。首先,MarkerMap可以扩展到大型数据集,而不需要特别的基因修剪。其次,它为监督学习和非监督学习提供了一个联合设置。第三,它是生成的,允许从减少的,信息数量的标志物插入到整个转录组水平。作者们提供了一组指标来评估插值的质量和比较原始转录组的分布与他们的重建。第四,它的监督选项强大地容忍小的标志物错误分类率,这可能来自加工和细胞类型分配错误。将MarkerMap应用于真实数据,包括用不同技术检测的脐带血单个核细胞(CBMCs),小鼠胚胎发生的纵向样本,以及发育中的小鼠大脑单细胞基因表达资源。最后,标志物选择与更广泛的可解释机器学习文献之间存在着密切的联系。由于这两个群体都在迅速发展,因此越来越需要系统地比较新的和现有的方法,以了解它们的优点和局限性。为了满足这一需求,作者们将MarkerMap与现有的标志物选择方法和来自更广泛的可解释机器学习文献的相关方法进行基准测试。目前,MakerMap作为一个pip可安装包(名称为“markermap”)提供。
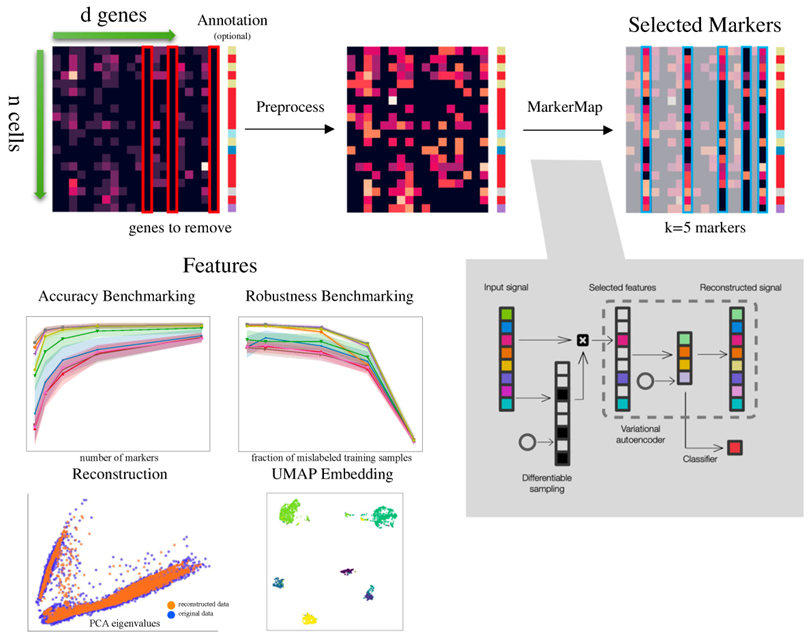
图1 MarkerMap计算管道。数据被导入为一个n × d的表达式计数数组,以及可选的注释。在预处理过程中,去除一些基因,其余基因进行缩放、归一化和log(1+x)变换。然后运行MarkerMap或各种其他标记选择算法来挑选k个标志物。这些标志物用于下游任务,包括基准测试、UMAP嵌入和数据重建。右下角描述了MarkerMap的体系结构。给定输入信号,可微采样过程选择一组全局标志物。在监督设置中,当注释可用时,限制于选定标志物的信号被馈送到预测标签的神经网络。在无监督版本中,限制于选择的信号被馈送到变分自编码器,其目的是在没有标签信息的情况下重建原始信号。联合损失版本使用重构损失和分类损失的凸组合。圆圈表示用于可微采样的随机输入源,可微采样是一种为信息特征迭代分配权重的技术。
参考文献
[1] Gregory W, Sarwar N, Kevrekidis G, Villar S, Dumitrascu B. MarkerMap: nonlinear marker selection for single-cell studies. NPJ Syst Biol Appl. 2024 Feb 14;10(1):17. doi: 10.1038/s41540-024-00339-3.
以往推荐如下:
5. EMT标记物数据库:EMTome
8. RNA与疾病关系数据库:RNADisease v4.0
9. RNA修饰关联的读出、擦除、写入蛋白靶标数据库:RM2Target
13. 利用药物转录组图谱探索中药药理活性成分平台:ITCM
19. 基因组、药物基因组和免疫基因组水平基因集癌症分析平台:GSCA
22. 研究资源识别门户:RRID
24. HMDD 4.0:miRNA-疾病实验验证关系数据库
25. LncRNADisease v3.0:lncRNA-疾病关系数据库更新版
26. ncRNADrug:与耐药和药物靶向相关的实验验证和预测ncRNA
28. RMBase v3.0:RNA修饰的景观、机制和功能
29. CancerProteome:破译癌症中蛋白质组景观资源
30. CROST:空间转录组综合数据库

https://blog.sciencenet.cn/blog-571917-1427059.html
上一篇:烟草PPO家族是参与传粉的重要调控因子
下一篇:数据库对理解lncRNA的贡献