博文
β-淀粉样蛋白和tau蛋白在阿尔茨海默病中的毒性交互作用
||
β-淀粉样蛋白和tau蛋白在阿尔茨海默病中的毒性交互作用
Amyloid‑β and tau — a toxic pas de deux in Alzheimer’s disease
澳大利亚悉尼大学神经精神疾病研究所 LarsM. Ittner Jürgen Götz
摘要:β-淀粉样蛋白(Aβ)和tau蛋白是阿尔茨海默病(AD)的标志性蛋白,针对其各自的毒性作用形式,已进行了广泛的研究。近期,两者在AD中可能的交互和协同效应已逐步得到阐明。本文将综述相关最新进展,对tau蛋白在AD发病中扮演角色的理解会转变为,tau蛋白是Aβ最重要的伙伴。对于tau蛋白细胞功能的理解也日渐深入,关注的焦点已从轴突(作为微管相关蛋白发挥首要作用)转移至树突(介导Aβ毒性)。
Ittner L,Götz J. Amyloid-β and tau-a toxic pas de deux in Alzheimer’s disease. Nat RevNeurosci, 2011, 12: 65-72
受全社会人口老龄化的影响,痴呆已成为重要的健康负担。2009年全球登记的阿尔茨海默病(AD)病例达到3560万,而估计到2050年这一数字将翻番。AD及相关类型痴呆仍无法治愈,目前的治疗方法只能达到中等程度的症状减轻[1,2]。
AD患者脑内除可见神经和突触丧失外,两项标志性的组织病理学损害为淀粉样斑块[含β-淀粉样蛋白(Aβ)]和神经原纤维缠结(NFT;微管相关蛋白tau的高度磷酸化形态)[3]。仅有tau病变,而无明显Aβ病变,为额颞痴呆(FTD;也称额颞叶变性,FTLD)亚型的特征;FTD是65岁以下人群第2位常见的痴呆类型。[4]。NFT病变进展并遍及全脑(该过程称“扩散”),且与AD进展相关[5];而突触丧失作为最早期事件,与患者的功能损害相关[6]。
在家族性AD(FAD)的少数亚型,已发现存在编码淀粉样前体蛋白(APP)、早老蛋白-1(PS1)或PS2的基因突变[7]。在家族性FTD的某亚型,携带编码tau蛋白(微管相关蛋白tau,MAPT)的基因突变[8],由此tau蛋白在神经变性疾病中的突出作用得以确立[8]。在散发性AD(SAD),载脂蛋白E4(APOE4)基因多态性及其他基因,也与发病风险增加相关[7]。AD和FTD患者中上述病理性突变的发现,有助转基因动物模型大量产生[9]。但需特别注意的是,绝大多数AD病例为散发性,其潜在病因仍不明,而目前的AD动物模型并非全面的人类疾病模型,仅模拟了疾病的关键方面。
近期针对AD的发病机制取得了实质性进展,如tau蛋白病变(及其他毒性蛋白)的神经元间扩散和跨脑区扩散[10]。并且人类研究、小鼠研究和体外研究已显示,Aβ与tau蛋白在AD中发挥毒性作用存在直接关联[11],但其交互作用的分子机制仍未阐明;十余年来,其一直是该领域的关键问题。本文将综述相关最新进展,包括Aβ与tau蛋白之间复杂的交互作用(特别是在突触部位)及其如何影响AD发病,而这些机制已日渐明朗。
Aβ对突触的毒性
APP经分步剪切后形成39-42个氨基酸残基的β-淀粉样肽[12,13]。Aβ易于聚集,产生毒性亚型,包括二聚体、寡聚体和纤丝[14]。虽然这三种Aβ形态中何种为毒性亚型尚存争议,但均认为突触(特别是突触后部)是Aβ毒性作用的主要靶点[6]。相应地,Aβ急性处理可致突触和树突棘丧失,多种实验范式下均可诱导长时程抑制(LTD)和长时程增强(LTP)[15]。虽然,突触后部可能仅存在单一受体介导Aβ毒性,但似乎多种突触后受体均有涉及,如朊毒体(prion)蛋白、α7-烟碱能受体、代谢型谷氨酸受体(mGluR),尤其是N-甲基-D-天冬氨酸受体(NMDAR)[15-17]。某特定受体介导的毒性,可能未必涉及Aβ与受体的直接结合,而是源自Aβ对受体属性的间接调控作用(可能是通过膜结合实现)。这便可以解释为何不同的实验设计中,Aβ在特定情况下仅结合于特定受体[18,19]。
有提示,过度刺激NMDAR引发的兴奋性毒性,可能为Aβ所致神经元损伤的核心机制,不过尚缺乏Aβ与NMDAR直接结合的证据[20]。有趣的是,NMDAR可介导Aβ诱导的树突棘丧失;而在同样实验条件下,mGluR介导Aβ诱导的LTD[15],提示不同受体介导Aβ毒性的不同方面。而进一步支持NMDAR在Aβ毒性中发挥作用的证据是,部分性NMDAR拮抗剂美金刚对AD患者有效[1]。
tau蛋白在轴突之外的新功能
tau蛋白包括有三个主要结构域:氨基端投射域(projectiondomain)、羧基端微管结合(MTB)重复序列,以及短尾部序列(shorttail sequence)。人脑中有六种tau蛋白亚型,通过外显子2、3(N-端插入序列)和外显子10的不同剪接方式而形成。外显子10可编码额外的MTB重复序列,生成有3个或4个MTB重复序列的亚型[8,21]。
tau蛋白主要在轴突发现,原因在于对其排列机制(sorting mechanism)并不完全清楚[22-25]。生理状态下,tau蛋白定位于树突,但其水平极低[26]。tau蛋白最明确的功能是稳定微管和调控动力驱动的轴突运输[27]。而tau蛋白与皮层膜的交互作用尚未完全了解[26,28]。进一步在神经元胞体-树突区(somatodendritic domain)发现了tau蛋白,是在病理状态下定位于此(下文详述)。
理论上,tau蛋白有84个磷酸化位点,在丝氨酸、苏氨酸和酪氨酸上分别有45个、35个和4个。tau蛋白磷酸化程度在神经元发育期较成熟期更高[29]。磷酸化对tau蛋白功能的影响所知尚少,但已知可负调控tau蛋白与微管的结合。在AD和FTD患者,以及tau蛋白转基因小鼠模型中,tau蛋白磷酸化程度增强(即高度磷酸化),同时见于生理性和病理性磷酸化位点(机制未完全阐明),导致其与微管分离[8]。因此认为tau蛋白的功能涉及到微管,如微管的稳定和轴突运输的调控[27];这些功能受损可能导致疾病。高度磷酸化tau蛋白在神经元胞体-树突区聚集、聚合,最终形成NFT[30]。有充分证据表明,可溶性的高度磷酸化tau蛋白在沉积之前,即可促成神经元功能紊乱[31]。进一步显示,高度磷酸化tau蛋白可干扰神经元功能,如影响线粒体呼吸和轴突运输等[27,32]。
tau蛋白是否为突触后支架蛋白?
tau蛋白通过其MTB重复序列与微管蛋白(tubulin)结合,而其投射区介导与其他蛋白[如酪氨酸蛋白激酶FYN[26,33]、动力蛋白激活蛋白(dynactin)[34]]的作用。dynactin与tau蛋白的交互作用,涉及到细胞内转运和(或)肌动蛋白(actin)与微管的耦联[34]。尽管早在1998年即体外发现FYN与tau蛋白交互作用,但其体内相关功能目前仍不清楚[33]。有趣的是,磷酸化tau蛋白和MAPT病理突变的表达产物,体外发现均可导致tau蛋白与FYN交互作用增强[35],提示其可能与疾病相关。FYN与tau的交互作用,在体内可易化FYN到突触后位点(即树突)的靶向(targeting)效应[该效应在tau蛋白敲除(tau-/-)小鼠中显著减少],随之FYN聚集于神经元胞体[26](图1)。在突触后部,FYN可磷酸化NMDAR 2B亚基(NR2B),从而介导NMDAR与突触后致密区蛋白95(PSD95)形成复合体[36]。而兴奋性毒性的下游信号转导,需要NMDAR与PSD95的交互作用[36]。而减少两者交互作用,tau-/-小鼠则降低针对实验性癫痫和Aβ毒性的易感性[20,26](图1)。
|
图1 破坏tau蛋白依赖性FYN树突靶向效应可保护AD小鼠模型神经元免受Aβ毒性 |
生理情况下,tau蛋白位于神经元的轴突和树突(后者水平较低)。酪氨酸蛋白激酶FYN位于树突区,可与tau蛋白交互作用;其还可磷酸化NMDA受体(NMDAR),由此介导后者与突触后致密区蛋白95(PSD95)的交互作用。而这种交互作用正是AD中β-淀粉样蛋白(Aβ)发挥毒性作用所必需,且在淀粉样前体蛋白(APP)转基因(APPtg)小鼠中导致兴奋性毒性、记忆受损和未成年死亡。增加tau蛋白水平的转基因小鼠(tautg)可见tau蛋白聚集于神经元胞体和树突,并伴突触后FYN水平增加。与此相关的是,双重转基因APPtg/tautg+小鼠(粗箭头)中Aβ毒性增加,较APPtg小鼠死亡率增加。截短的tau蛋白(Δtau)丧失微管结合特性,无法定位于树突,但可与FYN在神经元胞体交互作用。Δtau针对tau蛋白与FYN的交互作用发挥主导性的负性效应,从而防止FYN到达树突,得以保护APPtg/Δtau小鼠免受Aβ毒性。通过基因敲除技术删除tau蛋白(tau-/-),可防止tau蛋白依赖性FYN定位到树突,从而保护APPtg/tau-/-免受Aβ毒性。tau–/–和Δtau神经元胞体中FYN停留机制的差异,即可解释APPtg/tau-/-/Δtau小鼠中见到的额外保护效应。 |
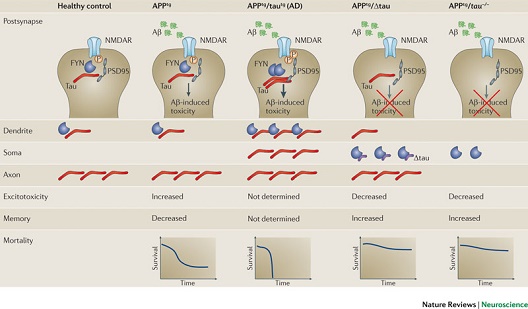
重要的是,强化免疫组化方法显示,生理状态的tau蛋白位于树突[26]。这一定位过程有赖于MTB重复序列(最可能是由于微管结合),因为缺乏MTB重复序列的截短tau蛋白(Δtau)并不结合于微管蛋白,活体中也不会到达树突[26](图1)。在tau蛋白介导的FYN靶向树突过程中,Δtau也会发挥主导性的负性效应,而在tau-/-模型中Δtau针对NMDAR介导信号转导发挥同样的效应,包括减轻Aβ毒性[26](图1)。微管在树突棘呈功能性存在(此为与突触可塑性相关的特征[37])的证据越来越多,继而tau蛋白在正常神经元的突触后部发挥重要的支架功能,这一可能性也大大增加。另一支持上述观点的证据是,tau蛋白与PSD95[26]之间存在强烈的交互作用,后者在NMDAR下游信号转导中即充当突触后支架蛋白的角色[36](图1)。总之,虽然tau蛋白主要见于轴突,但新发现的其树突功能对于正常神经元也极为重要,一旦受损即可能导致病变。
Aβ与tau蛋白的联系
按照淀粉样蛋白级联反应假说,Aβ的产生是造成AD病理变化的关键步骤[11]。支持该假说的证据为,FAD患者存在病理性突变,且与Aβ生成相关;还有21染色体三体者(携带额外的APP等位基因),其Aβ水平和AD发生率均较高[7]。接下来的关键问题就是,tau蛋白在淀粉样蛋白级联反应中处于何种地位?是Aβ毒性的首要靶点,中介物,还是某种意义上的旁观者?虽然Aβ和tau蛋白均可独立发挥毒性作用[38],但在体和离体模型均显示,两者间可能存在三种交互作用模式(图2)。
Aβ促进tau蛋白病变
Aβ作用于tau蛋白的模式呈现多个层次,这已有多方面的证据支持。在APP转基因小鼠,Aβ生成可致tau蛋白高度磷酸化,而在tau蛋白转基因小鼠,则并无Aβ斑块生成[39]。在这两种小鼠的杂交后代,可见NFT病变(而非Aβ斑块病变)加重[40,41](表1)。类似地,tau蛋白转基因小鼠脑内注射合成性Aβ,NFT病变同样加重[42]。进一步对三重转基因小鼠(3xTg-AD小鼠,Aβ斑块和NFT病变并见)进行抗Aβ免疫,则导致tau蛋白高度磷酸化程度降低[43]。有趣的是,近期研究显示,其他促淀粉样蛋白,如膜本体蛋白2B(integral membrane protein 2B;也称Bri肽,在英国和丹麦的家族性痴呆患者脑内聚集)也可以相同模式诱导NFT病变,提示NFT生成可由广泛的促淀粉样蛋白诱导,而不限于Aβ机制。
|
图2 β-淀粉样蛋白与tau蛋白:三种可能的交互作用机制 |
a Aβ通过造成tau蛋白高度磷酸化引起tau蛋白病变,进而介导对神经元的毒性。 b tau蛋白介导Aβ毒性,即Aβ毒性强烈依赖于tau蛋白的存在(树突的tau蛋白)。 c Aβ和tau蛋白针对细胞过程或细胞器发挥协同效应,从而放大各自的毒性效应。 |
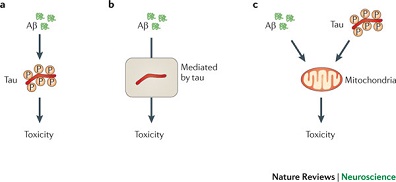
Aβ与tau蛋白具有毒性协同效应
Aβ和tau蛋白各自导致的下游毒性效应,分别针对同一系统的不同成分,从而放大了各自的效应。这种交互作用的典型就是AD小鼠模型的线粒体功能障碍,这一发病机制在神经变性中的作用被越来越多的发现[3]。在三重转基因小鼠,Aβ和tau蛋白均可损害线粒体呼吸,且并见Aβ斑块和tau蛋白病变[45]。有趣的是,tau蛋白倾向于损害呼吸链复合体Ⅰ,而Aβ阻断复合体Ⅳ依赖性线粒体呼吸,从而导致线粒体损害加重,两种病变叠加,不同于单一过表达tau蛋白或APP的小鼠[45]。
tau蛋白介导Aβ毒性
tau蛋白对Aβ毒性仅具次要作用的观点已受到实质性挑战,观察发现,培养的tau-/-神经元在Aβ诱导的细胞死亡过程中得以保存[46]。重要的是,这种保护作用经过两项独立的体内试验的重复验证(采用不同的APP转基因小鼠和tau蛋白缺陷小鼠[20,26])(表1)。其中一项研究显示,tau蛋白缺陷小鼠对Aβ诱导毒性的保护作用源于定位于树突的FYN减少,从而导致NMDAR与PSD95交互作用削弱,最终减轻了Aβ介导的兴奋性毒性[26]。要强调的是,不仅tau-/-或Δtau74小鼠(神经元表达Δtau)可对抗Aβ诱导毒性,某种治疗性肽也可通过破坏NMDAR-PSD95交互作用达到类似效应[26]。以上提示,tau蛋白依赖性树突信号转导在介导Aβ毒性中发挥关键作用。有趣的是,减少tau蛋白也可防止Aβ诱导的线粒体轴突运输受损[47];而tau蛋白正是通过这种运输方式介导Aβ毒性。
关于tau-/-小鼠的注释
首批tau-/-小鼠被报道时,令人吃惊的是并未见到明显的表型[48,49]。在某个tau-/-小鼠品系,轴突微管的微管蛋白排列仅轻微改变[48],而从另一个tau-/-小鼠品系获得的原代神经元细胞,其轴突生长也仅轻微延迟[49],原因可能在于微管相关蛋白1A(MAP1A)代偿了微管稳定作用[48]。而第三个tau缺陷小鼠品系获得的培养神经元,也可见类似的早期轴突形成延迟(L.M.I和J.G.未发表研究,在MAPT位点引入了绿色荧光蛋白[50])。有趣的是,老年(而非青年)tau-/-小鼠可见行为改变(如攻击和记忆受损),提示tau蛋白缺乏的代偿机制在早期尚可发挥作用,而晚期则可能失效[51]。这同样也能够解释APP转基因Tg2576小鼠与tau-/-小鼠杂交后代老年期的病理变化[52]。针对tau蛋白处理方式的长期效应是否导致了上述损害加重,尚有待证实。
表1 淀粉样蛋白-tau蛋白组合研究
年份 | 淀粉样蛋白模型 | tau蛋白模型 | 双重或三重转基因小鼠表型 | 参考文献 |
2001 | Tg2576(K670N/M671L‑APP695) | JNPL3(P301L‑(0N4R)tau) | (p‑tau,NFT) | 40 |
2001 | 合成性Aβ1‑42 | pR5(P301L‑(2N4R)tau) | (p‑tau,NFT) | 42 |
2003 | 3xTg AD(K670N/M671L‑APP695+M146V‑PS1) | (P301L‑(0N4R)tau) | Aβ和tau蛋白病变并见 | 62 |
2007 | APP23(K670N/M671L‑APP751) | 针对C57Bl/6小鼠JNPL3(P301L‑(0N4R)tau) | (p‑tau,NFT);晚发病变 | 63 |
2007 | J20(K670N/M671L/V717F‑APP751) | Tau–/– | 防止J20品系记忆损害和未成年死亡 | 20 |
2007 | J20(K670N/M671L/V717F‑APP751) | Tau–/– | 防止Aβ诱导的海马NPY表达和钙结合蛋白丧失 | 64 |
2008 | APP‑V717I(V717I‑APP695) | tau‑P301L(P301L‑(2N4R)tau) | (p‑tau),GSK3β活性增强 | 41 |
2010 | APP152(K670N/M671L‑APP751+N141I‑PS2) | pR5(P301L‑(2N4R)tau) | (p‑tau,NFT) | 65 |
2010 | Tg2576(K670N/M671L‑APP695) | Tau–/– | 老年小鼠记忆损害加重 | 52 |
2010 | ADnnPP7(795InsTTTAATTTGT‑BRI2) | TauP301S | (p‑tau,NFT) | 44 |
2010 | APP23(K670N/M671L‑APP751) | pR5(P301L‑(2N4R)tau) | 未成年死亡率增加 | 26 |
2010 | APP23(K670N/M671L‑APP751) | TauGFP/GFP(GFP敲入) | 防止APP23品系记忆丧失和未成年死亡 | 26 |
2010 | APP23(K670N/M671L‑APP751) | ∆tau74(1‑255aa tau) | 防止APP23品系记忆丧失和未成年死亡 | 26 |
Aβ:β-淀粉样蛋白;APP:淀粉样前体蛋白;GFP:绿色荧光蛋白;GSK3β:糖原合酶激酶3β;NFT:神经原纤维缠结;NPY:神经肽Y;p-tau:磷酸化tau蛋白;tg:转基因
AD的tau蛋白轴假说
基于有关tau蛋白的树突功能,及其介导Aβ毒性的新发现,为突出tau蛋白在疾病中的核心作用,作者提出新的“tau蛋白轴假说”,该假说可关联树突区的Aβ和tau蛋白病变。假说包含以下两部分:
第一,Aβ的突触后毒性为tau蛋白依赖性。更确切些,即tau蛋白与FYN交互作用,从而强化FYN到突触后部的靶向作用和(或)支架作用,FYN在突触后部连接NMDAR到下游信号通路(图1)。由此使NMDAR增敏,产生Aβ的毒性效应。这种tau依赖性Aβ毒性作用模式发生于神经元的树突区,涉及到兴奋性毒性信号转导。
第二,神经元暴露于Aβ(特别是持续性暴露)具有多种毒性作用。重要的是,Aβ进行性增强tau蛋白磷酸化(过度磷酸化)。由此导致tau蛋白与微管结合受损,造成异常神经元的胞体-树突区tau蛋白加速聚集(图3)。而且磷酸化tau蛋白与FYN亲和性也增强[35]。总之,以上的后果便是突触后FYN水平升高和NMDAR增敏,使得神经元树突对Aβ毒性更加易感。
|
图3 提议的阿尔茨海默病(AD)“tau蛋白轴假说”: 树突tau蛋白水平进行性升高增加神经元对Aβ的易感性 |
a AD(或轻度认知损害)的起病特征即为脑内Aβ的生成。然而,树突tau蛋白低水平(较之在轴突的高水平)导致神经元对突触(特别是突触后)Aβ毒性仅具有限的易感性。 b 随着疾病进展,tau蛋白磷酸化程度增加(受Aβ驱动),并聚集于神经元胞体-树突区,导致树突tau蛋白水平进行性升高。 c AD症状表现充分时,树突区高水平的tau蛋白使神经元对突触后Aβ毒性的易感性增加。Aβ毒性增加强化了tau蛋白磷酸化及其在胞体-树突区的聚集,从而使突触对Aβ毒性的敏感性增加,由此形成恶性循环。 |
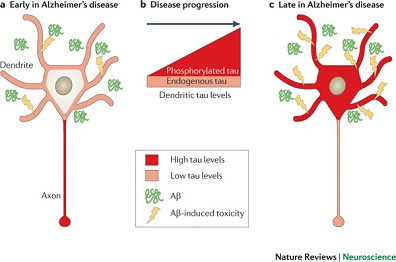
这样看来,淀粉样蛋白级联反应假说是聚焦于Aβ,而由于tau蛋白在树突新功能的明确,“tau蛋白轴假说”将这一关键性作用纳入。是否突触后Aβ毒性与tau蛋白磷酸化增强相关,进而形成恶性循环?尚有待确认。另外,虽然来自AD小鼠模型的证据提示,树突Aβ毒性是疾病的早期事件,但其在人类疾病中作用仍需明确。
前已述及,减少tau蛋白可防止Aβ诱导的线粒体轴突运输受损[47],这便将“tau蛋白轴假说”与以下两种假说联系起来,即轴突运输受损假说和氧化应激假说。前者认为因tau蛋白诱导而致轴突运输衰竭;后者认为线粒体(轴突运输的关键“货物”)功能受损导致活性氧的生成[55]。
未来方向
尽管tau蛋白的作用及其与Aβ的交互作用正日渐明朗,但人脑中6种tau蛋白亚型的功能仍是有待明确的问题之一。在小鼠中发现,位于树突的tau蛋白具有支架功能,提示这一部位和(或)其他细胞区室可能有多种tau蛋白的支架蛋白伙伴,且不同tau蛋白亚型可能会形成不同的复合物。进一步讲,tau蛋白在细胞质膜中的功能,与其他蛋白联系并最终靶向特定亚细胞区室的机制,均尚未完全阐明[28]。
培养神经元的Aβ毒性同时具有tau蛋白依赖性和FYN依赖性[46,56]。其毒性与特定Aβ亚型聚集,并募集tau蛋白至脂筏(lipid raft)相关联[56]。这可能提示,活体内的脂筏可与Aβ和tau蛋白在特定的膜区室相联系。进一步还需明确是否FYN的变化对tau蛋白有直接影响。
针对tau蛋白依赖性机制已成为治疗tau蛋白病变(包括AD[2])的适宜策略(治疗AD似需谨慎采用同时针对tau蛋白和Aβ的组合手段)。就tau蛋白来讲,降低其水平、破坏NMDAR-PSD95或Fyn-Tau交互作用,都是合理的治疗选择[26]。鉴于多数证据提示只有磷酸化tau蛋白才具有毒性[57],降低激酶活性(如糖原合酶激酶-3)[58,59],或增加磷酸酶活性(如丝氨酸/苏氨酸蛋白磷酸酶2A)[60],对转基因AD小鼠模型均显示有效,有可能会转化为临床应用。而上述策略降低tau蛋白生理水平的同时,尚需观察是否会对各型细胞产生副作用(如在少突胶质细胞中阻止tau蛋白依赖性FYN定位,可影响髓鞘形成[61])。
一百多年前,阿洛伊斯·阿尔茨海默在显微镜下发现淀粉样斑块和NFT的时候,他是否会预见到形成这些病变的蛋白会有如此复杂的功能,又会与疾病的发生有如此紧密的联系呢?
1. Citron, M. Strategies for diseasemodification inAlzheimer’s disease. Nature Rev. Neurosci. 5, 677–685 (2004).
2. Brunden, K. R., Trojanowski, J. Q.& Lee, V. M.Advances in tau‑focuseddrug discovery for Alzheimer’sdisease and related tauopathies. Nature Rev.DrugDiscov. 8, 783–793 (2009).
3. Querfurth, H. W. & LaFerla, F. M.Alzheimer’s disease.N. Engl. J. Med. 362, 329–344 (2010).
4. Cairns, N. J. et al. Neuropathologicdiagnostic andnosologic criteria for frontotemporal lobardegeneration:consensus of the Consortium forFrontotemporal Lobar Degeneration.ActaNeuropathol. 114, 5–22 (2007).
5. Braak, H. & Braak, E. Staging ofAlzheimer’s disease‑
related neurofibrillary changes.Neurobiol. Aging 16,271–278; discussion 278–284 (1995).
6. Selkoe, D. J. Alzheimer’s disease is asynaptic failure.Science 298, 789–791 (2002).
7. Bertram, L. & Tanzi, R. E. Thegenetic epidemiology ofneurodegenerative disease. J. Clin. Invest.115,1449–1457 (2005).
8. Ballatore, C., Lee, V. M. &Trojanowski, J. Q. Tau‑
mediated neurodegeneration in Alzheimer’sdiseaseand related disorders. Nature Rev. Neurosci. 8, 663–672 (2007).
9. Götz, J. & Ittner, L. M. Animalmodels of Alzheimer’sdisease and frontotemporal dementia. Nature Rev.Neurosci.9, 532–544 (2008).
10. Frost, B. & Diamond, M. I. Prion‑like mechanismsinneurodegenerative diseases. Nature Rev. Neurosci.11, 155–159 (2010).
11. Hardy, J. & Selkoe, D. J. Theamyloid hypothesis ofAlzheimer’s disease: progress and problems on theroad totherapeutics. Science 297, 353–356 (2002).
12. Sisodia, S. S. & St George‑Hyslop, P. H. γ‑Secretase,notch, Aβand Alzheimer’s disease: where do thepresenilins fit in? Nature Rev. Neurosci.3, 281–290(2002).
13. LaFerla, F. M., Green, K. N. &Oddo, S. Intracellularamyloid‑β inAlzheimer’s disease. Nature Rev.Neurosci. 8, 499–509 (2007).
14. Haass, C. & Selkoe, D. J. Solubleprotein oligomers inneurodegeneration: lessons from the Alzheimer’samyloid β‑peptide. NatureRev. Mol. Cell Biol. 8, 101–112 (2007).
15. Shankar, G. M. et al. Amyloid‑β proteindimersisolated directly from Alzheimer’s brains impairsynaptic plasticity andmemory. Nature Med. 14,837–842 (2008).
16. Lauren, J., Gimbel, D. A., Nygaard, H.B., Gilbert, J. W.& Strittmatter, S. M. Cellular prion proteinmediatesimpairment of synaptic plasticity by amyloid‑βoligomers. Nature457, 1128–1132 (2009).
17. Snyder, E. M. et al. Regulation ofNMDA receptortrafficking by amyloid‑β.Nature Neurosci. 8, 1051–1058 (2005).
18. Kessels, H. W., Nguyen, L. N., Nabavi,S. & Malinow,R. The prion protein as a receptor for amyloid‑β.Nature 466, e3–e4(2010).
19. Small, D. H. et al. The β‑amyloid protein ofAlzheimer’sdisease binds to membrane lipids but does not bind tothe α7nicotinic acetylcholine receptor. J. Neurochem.101, 1527–1538 (2007).
20. Roberson, E. D. et al. Reducingendogenous tauameliorates amyloid β‑induceddeficits in anAlzheimer’s disease mouse model. Science 316, 750–754 (2007).
21. Goedert, M. & Spillantini, M. G. Acentury ofAlzheimer’s disease. Science 314, 777–781 (2006).
22. Hirokawa, N., Funakoshi, T., Sato‑Harada, R. &Kanai,Y. Selective stabilization of tau in axons andmicrotubule‑associated protein2C in cell bodies anddendrites contributes to polarized localizationofcytoskeletal proteins in mature neurons. J. Cell Biol.132, 667–679 (1996).
23. Aronov, S., Aranda, G., Behar, L.& Ginzburg, I. Axonal tau mRNA localization coincides with tauprotein inliving neuronal cells and depends on axonal targeting signal. J. Neurosci. 21,6577–6587(2001).
24. Utton, M. A. et al. The slow axonaltransport of themicrotubule‑associatedprotein tau and the transport rates of different isoforms and mutantsincultured neurons. J. Neurosci. 22, 6394–6400(2002).
25. Konzack, S., Thies, E., Marx, A.,Mandelkow, E. M. &Mandelkow, E. Swimming against the tide: mobility ofthemicrotubule‑associatedprotein tau in neurons.J. Neurosci. 27, 9916–9927 (2007).
26. Ittner, L. M. et al. Dendriticfunction of tau mediatesamyloid‑βtoxicity in Alzheimer’s disease mousemodels. Cell 142, 387–397 (2010).
27. Götz, J., Ittner, L. M. & Kins, S.Do axonal defects intau and amyloid precursor protein transgenic animalsmodelaxonopathy in Alzheimer’s disease?J. Neurochem. 98, 993–1006 (2006).
28. Maas, T., Eidenmuller, J. &Brandt, R. Interaction oftau with the neural membrane cortex is regulatedbyphosphorylation at sites that are modified in pairedhelical filaments. J.Biol. Chem. 275, 15733–15740(2000).
29. Brion, J. P., Smith, C., Couck, A. M.,Gallo, J. M. &Anderton, B. H. Developmental changes in tauphosphorylation:fetal tau is transientlyphosphorylated in a manner similar to pairedhelicalfilament‑taucharacteristic of Alzheimer’s disease.J. Neurochem. 61, 2071–2080 (1993).
30. Götz, J. et al. Somatodendriticlocalization andhyperphosphorylation of tau protein in transgenicmiceexpressing the longest human brain tau isoform.EMBO J. 14, 1304–1313 (1995).
31. Santacruz, K. et al. Tau suppressionin aneurodegenerative mouse model improves memoryfunction. Science 309, 476–481(2005).
32. Ittner, L. M., Ke, Y. D. & Götz,J. Phosphorylated Tauinteracts with c‑JunN‑terminal kinase‑interactingprotein1 (JIP1) in Alzheimer disease. J. Biol. Chem.284, 20909–20916 (2009).
33. Lee, G., Newman, S. T., Gard, D. L.,Band, H. &Panchamoorthy, G. Tau interacts with src‑family non‑
receptor tyrosine kinases. J. Cell Sci.111, 3167–3177(1998).
34. Magnani, E. et al. Interaction of tauprotein with the dynactin complex. EMBO J. 26, 4546–4554(2007).
35. Bhaskar, K., Yen, S. H. & Lee, G.Disease‑relatedmodificationsin tau affect the interaction between Fynand Tau. J. Biol. Chem. 280,35119–35125 (2005).
36. Salter, M. W. & Kalia, L. V. Srckinases: a hub forNMDA receptor regulation. Nature Rev. Neurosci. 5,317–328(2004).
37. Gu, J. & Zheng, J. Q. Microtubulesin dendritic spinedevelopment and plasticity. Open Neurosci. J. 3,128–133(2009).
38. Small, D. H., Mok, S. S. &Bornstein, J. C. Alzheimer’sdisease and Aβ toxicity: from top to bottom.NatureRev. Neurosci. 2, 595–598 (2001).
39. Götz, J. et al. Transgenic animalmodels of Alzheimer’sdisease and related disorders: histopathology,behavior andtherapy. Mol. Psychiatry 9, 664–683(2004).
40. Lewis, J. et al. Enhancedneurofibrillary degenerationin transgenic mice expressing mutant tau andAPP.Science 293, 1487–1491 (2001).
41. Terwel, D. et al. Amyloid activatesGSK‑3βtoaggravate neuronal tauopathy in bigenic mice. Am.J. Pathol. 172, 786–798(2008).
42. Götz, J., Chen, F., van Dorpe, J.& Nitsch, R. M.Formation of neurofibrillary tangles in P301L tautransgenicmice induced by Aβ42 fibrils. Science 293,1491–1495 (2001).
43. Oddo, S., Billings, L., Kesslak, J.P., Cribbs, D. H. &LaFerla, F. M. Aβ immunotherapy leads to clearanceofearly, but not late, hyperphosphorylated tauaggregates via the proteasome.Neuron 43, 321–332(2004).
44. Coomaraswamy, J. et al. Modelingfamilial Danishdementia in mice supports the concept of the amyloidhypothesisof Alzheimer’s disease. Proc. Natl Acad.Sci. USA 107, 7969–7974 (2010).
45. Rhein, V. et al. Amyloid‑β and tausynergisticallyimpair the oxidative phosphorylation system in tripletransgenicAlzheimer’s disease mice. Proc. Natl Acad.Sci. USA 106, 20057–20062 (2009).
46. Rapoport, M., Dawson, H. N., Binder,L. I., Vitek, M. P.& Ferreira, A. Tau is essential to β‑amyloid‑inducedneurotoxicity.Proc. Natl Acad. Sci. USA 99, 6364–6369 (2002).
47. Vossel, K. A. et al. Tau reductionprevents Aβ‑induceddefectsin axonal transport. Science 330, 198 (2010).
48. Harada, A. et al. Altered microtubuleorganization insmall‑calibreaxons of mice lacking tau protein. Nature369, 488–491 (1994).
49. Dawson, H. N. et al. Inhibition ofneuronal maturation in primary hippocampal neurons from tau deficient mice. J.Cell Sci. 114, 1179–1187 2001).
50. Tucker, K. L., Meyer, M. & Barde,Y. A. Neurotrophinsare required for nerve growth during development.NatureNeurosci. 4, 29–37 (2001).
51. Takashima, A. The mechanism for tauaggregation andits relation to neuronal dysfunction. Alzheimer’s &Dementia6, S144 (2010)
52. Dawson, H. N. et al. Loss of tauelicits axonaldegeneration in a mouse model of Alzheimer’s disease.Neuroscience169, 516–531 (2010).
53. Saper, C. B., Wainer, B. H. &German, D. C. Axonal andtransneuronal transport in the transmissionofneurological disease: potential role in systemdegenerations, includingAlzheimer’s disease.Neuroscience 23, 389–398 (1987).
54. Stamer, K., Vogel, R., Thies, E.,Mandelkow, E. &Mandelkow, E. M. Tau blocks traffic of organelles,neurofilaments,and APP vesicles in neurons andenhances oxidative stress. J. Cell Biol. 156,1051–1063 (2002).
55. Pappolla, M. A., Omar, R. A., Kim, K.S. & Robakis,N. K. Immunohistochemical evidence of oxidative[corrected]stress in Alzheimer’s disease. Am.J. Pathol. 140, 621–628 (1992).
56. Williamson, R., Usardi, A., Hanger, D.P. & Anderton,B. H. Membrane‑bound β‑amyloid oligomersarerecruited into lipid rafts by a fyn‑dependentmechanism.FASEB J. 22, 1552–1559 (2008).
57. Ittner, L. M. et al. Parkinsonism andimpaired axonaltransport in a mouse model of frontotemporal dementia.Proc. NatlAcad. Sci. USA 105, 15597–16002 (2008).
58. Gong, C. X. & Iqbal, K.Hyperphosphorylation ofmicrotubule‑associatedprotein tau: a promisingtherapeutic target for Alzheimer disease. Curr.Med.Chem. 15, 2321–2328 (2008).
59. Noble, W. et al. Inhibition ofglycogen synthasekinase‑3 bylithium correlates with reduced tauopathyand degeneration in vivo. Proc. NatlAcad. Sci. USA102, 6990–6995 (2005).
60. van Eersel, J. et al. Sodium selenatemitigates taupathology, neurodegeneration, and functional deficitsinAlzheimer’s disease models. Proc. Natl Acad. Sci.USA 107, 13888–13893 (2010).
61. Klein, C. et al. Process. outgrowth ofoligodendrocytesis promoted by interaction of fyn kinase with thecytoskeletalprotein tau. J. Neurosci. 22, 698–707(2002).
62. Oddo, S. et al. Triple‑transgenic model ofAlzheimer’sdisease with plaques and tangles: intracellular Aβ and synapticdysfunction. Neuron 39, 409–421 (2003).
63. Bolmont, T. et al. Induction of taupathology byintracerebral infusion of amyloid‑β‑containingbrainextract and by amyloid‑βdeposition in APP x Tautransgenic mice. Am. J. Pathol. 171, 2012–2020 (2007).
64. Palop, J. J. et al. Aberrantexcitatory neuronal activityand compensatory remodeling ofinhibitoryhippocampal circuits in mouse models of Alzheimer’sdisease. Neuron55, 697–711 (2007).
65. Grueninger, F. et al. Phosphorylationof Tau at S422 isenhanced by Aβ in TauPS2APP triple transgenic mice.Neurobiol.Dis. 37, 294–306 (2010).
β-淀粉样蛋白与tau蛋白在阿尔茨海默病中的毒性交互作用(Amyloid?β and tau — a to.pdf
https://blog.sciencenet.cn/blog-426290-906127.html
上一篇:衰老过程中端粒、线粒体及干细胞功能衰退的相关性
下一篇:美国老年医学的未来 The Future of Geriatric Medicine
全部作者的精选博文
全部作者的其他最新博文
全部精选博文导读
相关博文
- • 聚英才 建高地 | 北京理工大学“特立青年学者”全球招聘开启
- • 700年后日本或濒临灭绝?日本学者推算预测:届时或仅剩1名15岁以下孩子
- • [转载]【同位素视角】非英语母语学者如何区分’e.g.’, ‘i.e.’, ‘namely’与‘such as’等混淆难题
- • 美国佐治亚大学等机构学者:刈割策略对Bulldog 805紫花苜蓿+Tifton 85狗牙根混播草地产量及品质的影响
- • 美国堪萨斯州立大学、密苏里大学等机构学者研究成果:土壤水分管理策略和品种多样性对紫花苜蓿产量、营养品质和农场盈利能力的影
- • 德国、捷克草业科学学者长期放牧实验:异质草地斑块中的土壤有机碳储量和地下生物量