博文
RNA病毒基因组组装指南
 精选
精选
|
从前几年的猪流感和埃博拉,再到上个月在韩国流行的MERS, 病毒的每次爆发都能使全球陷入一阵恐慌,病毒虽然没有真正在全球爆发,但是各国在预防上消耗的资源比在治疗上消耗的还要多。殊不知,病毒是世界上最简单的生物,简单到到很多人甚至不愿意把他们归为生物这一类,那为什么这么简单的病毒,每年冬天都会让那么多人得流感呢?
你也许会说,病毒也没那么可怕吧,人类不是成功地把天花病毒从地球上抹去了吗?这倒是,但是为什么人类却总不能消灭流行性感冒呢?这得从病毒的基因组结构说起,天花病毒是一种DNA病毒,也就是说它的基因组结构是DNA,而第一段里提到的三种病毒的基因组却恰恰都是RNA构成的。它们的区别就在于,DNA病毒不容易变异,往往一次接种就可以管用几十年,而RNA病毒却变异很快,往往过了一年,去年接种的疫苗在变异之后的病毒身上已经不管用了,这就是为什么每年都会有上千万的人得流感的原因了。
在抗击这种可以随时变异的病毒来说,对它进行基因组测序是一个相当好的办法,测序不仅能够让我们很快地追溯到变异的来源,而且也能帮助估计它对人体的危害程度和现有疫苗的有效性。这次五月份MERS爆发,中国疾控中心在MERS从韩国传入中国后的第二天就测出了MERS的基因组,而韩国花了将近三周时间才把MERS的基因组公布出来。
虽然病毒的基因组相当小,一般也就一万个碱基对左右,可是对它的的测序却也没那么简单。有时候实验室为了取得活体样品,需要千里迢迢赶到样品所在地,通过层层海关才能把样品运回来,毕竟带着病毒跑来跑去也是很严肃的事情。而且想把病毒养在培养的细胞里也并不容易。另外,因为病毒只有在宿主细胞里才能存活,所以提取病毒RNA测序之后结果里往往会混有宿主的RNA,如果直接对组织提取RNA的话,病毒的RNA量有时只能占到总RNA的百分之一,而且有时候在宿主的一块组织里有不止一种病毒,这导致几种病毒的基因组片段也会混在一起,这都会对后期的组装造成一些不小的麻烦。那么如何从这些混杂的序列里找出你想要的基因组呢?
测序之后接下来要做的就是数据处理,既然在通常情况下测序数据里会混有宿主的基因组片段,那么在组装之前就需要先去掉这些宿主来源的序列。基本的思路是,将所有的reads都对比到宿主的基因组上,然后从原本的reads里去掉那些可以被对比到宿主基因组上的reads。如果宿主的基因组无法得到的话,也可以找一个亲缘关系比较近的物种的基因组,然后将匹配的标准稍微降低一点。因为是RNA测序,mapping软件应该用适合RNA测序的,具体原因可以参考我的RNA测序的介绍,我用的一直是Tophat,效果还算不错。在用Tophat对比完之后生成的SAM文件里,就包含了所有匹配和没匹配到基因组上的reads的信息,Tophat的好处是会把发现匹配的和没匹配的reads分成两个SAM文件。SAM文件的第一行的reads的名称和原来fastq文件夹里的reads名称是相同的,所以根据reads的名称就可以把SAM文件和原始的fastq文件关联起来。(如果其他对比软件不自动把两类SAM文件分来的话,根据SAM文件的第四行也可以轻松辨别一个read是不是在基因组上发现了匹配。没有匹配上的话是这一列会是0,对比上的话这里就会是一个大于0的数)
知道了宿主来源的reads的名字之后,就可以在原始的fastq文件里查找并且删除了,对于每一个宿主来源的reads名称,我们可以遍历整个fastq文件来查找匹配到的reads然后把它删除(如果是paired-end reads的话就需要同时把两个fastq文件里相对应的reads都删掉),但是这样做会很慢,操作次数基本上是两个集合元素个数的乘积。所以可以先对两个reads名字的集合进行排序,因为匹配到的reads的名字的集合一定是所有的reads名字集合的一个子集,排序之后,我们只需要将总reads集合里的每一个元素始终和小集合里第一个元素进行比较,如果发现相同,就把小集合里的第一个元素删掉,后面的推上来。这样的话就可以在遍历一遍reads的情况下标记下所有map上去的reads,计算量就只是落在对两个集合排序上了,而排序所需要的操作次数比之前挨个查找的方法减少了几千倍。这个python小程序可以读取两个paired-end fastq文件和一个SAM文件,然后用这种方式输出两个去除宿主来源之后的paired-end fastq文件。
但有时会出现的情况是,病毒序列里混着的不只是一种宿主的序列片段。这时候就可以再用一次tophat对另外一个宿主的基因组进行mapping,然后将两个不同宿主的mapping的SAM文件合并,再进行上面的操作就可以了。
当我们得到了相对纯净的reads,下一步就可以用软件来组装了。和大基因组相比,病毒的基因组一般很小,这样对组装软件所占的内存就不需要特别在意,而且病毒的基因组基本上没有重复,所以说对算法的优化程度的要求也不高。组装软件有很多,一般基于三种算法。一种是比较直观的贪婪字符串延长算法 (string-based-algorithm, SBA),每次都找到最好的重叠然后把他们合并成一条,直到不能合并为止。而现在用得比较多两种是图算法,分别叫做overlap-layout-consensus (OLC) 和 de-Bruijn-graph (DBG),它们是通过寻找reads之间的重叠或者将reads分成更小的片断来作出一个有顶点和箭头的图,在图中的顶点都表示一段序列,箭头表示他们之间的重叠或者相连。最后在画好的图里找到一个或者几个连接这些顶点的最佳路径,每条路径就代表一个拼接好的长片段,或者叫contigs,病毒的基因组很有可能就已经在这些contigs里面了。我比较了几种最受欢迎的小基因组拼接软件在我的数据上的效果。其中的N50和N90是一般评价拼接软件的一个指标,指如果把所有的contigs按长度由大到小拼在一起之后,在总长度的50%和90%位置的那个contig的长度。当然这个指标有点肤浅,没有考虑到contigs的可靠性和覆盖度,不过对简单的比较来说就已经足够了。
EDena | velvet | Minia | Allpaths-lg | Taipan | Mira | |
算法 | OLC | DBG | DBG | DBG | SBA&GBA | OLC/DBG |
可用性 | Failed | Pass | Pass | not-suitable | not-suitable | Pass |
N50 (bp) | 370 | 967 | 297 | NA | NA | 835 |
N90 (bp) | 261 | 286 | 164 | NA | NA | 334 |
contig总数 | 53 | 641 | 3396 | NA | NA | 19190 |
>2500 bp | 0 | 18 | 12 | NA | NA | 411 |
时间 | <30min | <30min | <30min | NA | NA | >15h |
Minia是我用的第一个软件,是一个非常简单易用的软件,速度也比较快,但是Minia的缺点在于它不报告组装好的contigs的coverage,也不能设定输出的contigs的最小长度,这样在后来检查contigs的可靠性上就不是那么方便。
Edena在安装使用起来也很方便, 但是它不支持在一个文件里的reads长短不一的情况,对于我们的数据,因为去除了很多adapter和一些质量不好的部分,所以只有不到一半还是全长,用那些不到一半的数据拼接的效果可以说惨不忍睹,最长的一个contig只有1076bp。而Minia, Mira和Velvet都拼接出了我10000多bp长的对照病毒的基因组。
ALLPATHS-LG是broad institute和剑桥一起开发的一个序列组装软件,在评测文章里对小基因组和大基因组的拼接表现都比较突出。可是好的表现是用更多的工作量换来的,ALLPATHS-LG组装需要至少两个library, 一个在理论上有少部分重叠的短片断library和一个大片段的library,致使实验操作和后期分析都比较麻烦。因为我只有一个libary,而且是不重叠的,所以ALLPATHS-LG不能用在这种数据上,ALLPATHS-LG同样也不支持454和sanger的测序数据。
Taipan是唯一的一个同时利用string-based算法和图算法的拼接软件,但是Taipan也需要输入文件里的reads都是一样的长度,而且Taipan的输入格式是RAW格式,也就是文件里除了序列什么都不提供,这样的话就没有利用Fastq文件里对应于每个碱基的测序质量信息,也当然没有用到pair-end的信息,而且Taipan只能拼接非常非常短的序列,对于我两头各161bp的reads已经无能为力了,所以说Taipan还需要改进很多。
Mira是德国癌症研究中心(DKFZ)开发的,是到目前为止最友好的序列组装软件。它不仅有自己的windows用户界面,而且作者为它洋洋洒洒写了200页的使用手册,要知道程序员最不喜欢的两件事情,一是写手册,二是别人不写手册,这跟Taipan两页的手册比起来有诚意不少。Mira在组装小于一亿个bp的基因组时比较有效,但是操作却没有Velvet简便,需要写一个说明文件给Mira。另外Mira的组装时间明显比其他几个软件要长很多,最后的记录文件都能有300多兆,感觉Mira作者实在是和唐僧有得一拼。。。
Velvet是从我一开始做别人就推荐我用的,但我还是想试试其他的软件,试了一圈最后还是发现Velvet好用。它的优点是速度快,对每个contig都报告覆盖度,而且可以指定只输出大于一定长度的contigs. 我的测试数据里有三种病毒的基因组片断,Velvet也是一点也不费力地全部拼接出来了。
找到了contigs之后要做的就是验证找到的contigs里有没有想要的RNA病毒的基因组。如果contigs少的话,就可以直接复制粘贴到数据库里检索;如果contigs很多的话,可以把这些contigs按照相似度分类,然后找到每个分类里最长的那一条。这里有一个python小程序可以对contigs文件里的每两个contigs进行局部相似度对比,这个对比是基于Biopython里一个经过修改的动态规划算法,两条序列的长度并不会对他们之间的局部相似度造成影响。其中动态规划的具体参数,比如缺口罚分和匹配奖分,可以根据所有序列的整体相似度在程序里调试。这个程序的输出是一个相似度矩阵,根据这个相似度矩阵做一个简单的聚类分析就可以大概确定contigs之间的关系了,就像下面这张图:
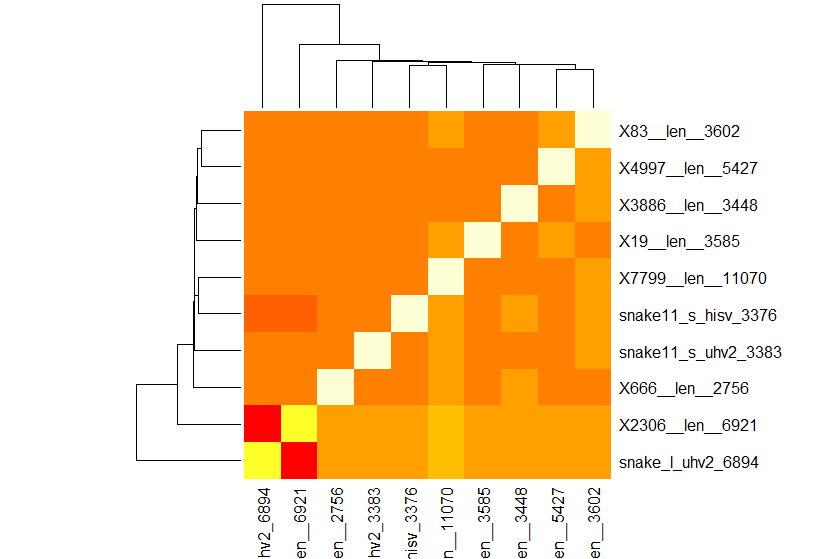
在图里有三个病毒的基因组,最下面两条序列分别是两个病毒基因组的长链,倒数第四和第五是这两个病毒基因组的短链,再往上一位是一个单独的单链病毒。在根据相似度分类以后,这三个病毒的基因组之间的关系就比较明显了。所以在得到contigs以后,可以把可信度比较高的contigs和一些已知的亲缘关系和近的病毒的基因组做比较,来确定这个contig是不是某个病毒的基因组了。
到目前为止,在GOLD基因组数据库里已经记录完成了四万多个细菌和将近一万个真核生物的基因组测序项目,可是却只有四千多个完成的病毒基因组测序项目。这和病毒基因组的测序难度完全不成正比。这或许是因为病毒基因组的研究还没有被病毒学家们重视起来。人类虽然成功消灭过天花和牛瘟病毒,但对很多病毒的袭击仍旧束手无策。最近几年的禽流感,虽然致死率低,但是很容易传播,埃博拉虽然不容易传播,但是致死率却相当高,如果以后出现一种又容易传播并且致死率高的病毒,我们仍旧有信心打败它吗?
https://blog.sciencenet.cn/blog-635619-902677.html
上一篇:用tophat和cufflinks分析RNAseq数据
下一篇:用哈希高效实现NGS序列的k-mer检测